


Hypoplastic left heart syndrome is characterised by failure of development of the left ventricle often in association with hypoplasia or atresia of the mitral and aortic valves. Although there are several underlying genetic mutations or abnormalities associated with hypoplastic right heart syndromes, there are few underlying genetic abnormalities associated with hypoplastic left heart syndrome. We describe a case of a baby girl born with hypoplastic left heart syndrome with hypoplastic branch pulmonary arteries who was found to have 22q11.21 microduplication on fluorescence in situ hybridisation and array comparative genomic hybridisation analysis.
Case report
The proband, a baby girl, was born at 38 weeks of gestation via lower segment caesarean section to healthy non-consanguineous Caucasian parents. Her birth weight was 2.55 kg. This was the couple’s third pregnancy, the previous two resulting in first trimester miscarriages. All three pregnancies resulted from ovarian stimulation. Foetal echocardiography at 20 weeks of gestation showed a hypoplastic left heart with mitral and aortic atresia.
The patient presented on the 1st day of life with cyanosis, with an oxygen saturation measuring 75%. She had weak femoral and brachial pulses. There was a prominent right ventricular impulse with a prominent single second heart sound. There was no audible murmur. Electrocardiogram demonstrated a right axis and right ventricular hypertrophy. Echocardiography demonstrated hypoplastic left heart syndrome consisting of mitral and aortic atresia, severe hypoplasia of the ascending aorta measuring 1.5 mm, and severe left ventricular hypoplasia. There was a high secundum atrial septal defect with left-to-right shunting. The aortic arch was left sided with ductal shunting from right to left with retrograde flow around the ascending aorta. Of note, the pulmonary arteries were hypoplastic, measuring 3 mm. There was mild tricuspid regurgitation and normal right ventricular function. The child was commenced on prostaglandin at 5 ng/kg per minute. Cranial and renal ultrasound scans were normal. Fluorescence in situ hybridisation analysis revealed a microduplication of chromosome 22q11.2. Subsequent array comparative genomic hybridisation analysis confirmed chromosome 22q duplication at band 22q11.21. The proband exhibited subtle dysmorphic features (Fig 1) including brachycephaly, a high forehead with mild bitemporal narrowing, slightly upslanting palpebral fissures, depressed nasal bridge, prominent nasal tip, relatively flat philtrum, thin lips, downturned corners of the mouth, a right ear pit, and a wide internipple distance. She had mild speech delay.
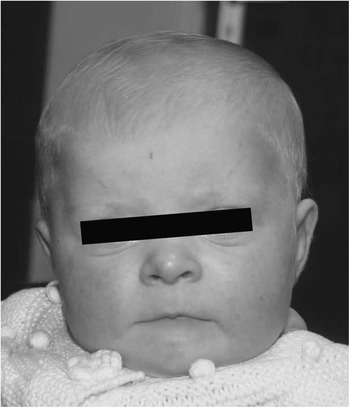
Figure 1 Brachycephaly, high forehead with bitemporal narrowing, mildly upslanting palpebral fissures, depressed nasal bridge, prominent nasal tip, long and relatively flat philtrum, thin lips, and downturned corners of the mouth.
At 5 days of age, she underwent a Norwood procedure with Sano shunt. She had an uncomplicated procedure and was extubated 4 days later and discharged home 3 weeks after surgery despite the hypoplastic pulmonary arteries (Fig 2a). A Glenn shunt was performed at 4 months of age. She had an uncomplicated post-operative course. A catheterisation was performed at 2 years of age after her Glenn shunt. This demonstrated pulmonary arterial pressures, which were slightly elevated with a mean pressure of 15 mmHg and pulmonary vascular resistance of 2.9 Wood units. She was treated with sildenafil, bosentan, and oxygen.

Figure 2 Angiogram demonstrating hypoplastic branch pulmonary arteries following Sano procedure ( a ) and significant growth in the pulmonary arteries following bi-directional Glenn ( b ).
Repeat catheterisation at 3 years of age demonstrated a mean pulmonary arterial pressure of 11 mmHg. A stent was placed in the left pulmonary artery, which was mild to moderately hypoplastic (Fig 2b), and collateral vessels were coil occluded. Her oxygen saturation was 72% in room air. She underwent a fenestrated extracardiac total cavopulmonary connection 3 months later. She was extubated within 48 hours, weaned off inotropes by day 4, and the chest drains were removed on day 9 post operation. She was discharged home 19 days post Fontan on enalapril, diuretics, warfarin, and pulmonary vasodilators but not on oxygen. Her oxygen saturation in room air was 84% on discharge.
Discussion
We report the case of a child with hypoplastic left heart syndrome with hypoplastic pulmonary arteries, mildly dysmorphic craniofacial features, and speech delay in association with a microduplication of chromosome 22q11.21. Fluorescence in situ hybridisation analysis with a probe mapping to the region (D22S75, Kreatech) showed duplication chromosome 22q11.2 on interphase nuclei. Subsequent array comparative genomic hybridisation testing, using an oligonucleotide array with ~60,000 probes across the genome at 300 kb gain and 100 kb loss resolution, showed a gain of ∼2.3 Mb in the long arm of chromosome 22 at band 22q11.21, between base pair coordinates 18,919,942 and 21,307,949, a region that includes the TBX1 gene. Fluorescence in situ hybridisation analysis on the parents showed a normal result for both, and thus the microduplication in the proband appears to have occurred as a de novo event, although germline mosaicism cannot be ruled out.
Chromosome 22, in particular band 22q11.2, has been shown to be susceptible to rearrangements due to misalignment of low copy repeats in this region. Microdeletions within this band in patients with DiGeorge/velocardiofacial syndrome were first described by Driscoll et alReference Driscoll, Budarf and Emanuel 1 in 1992. Well-reported features of DiGeorge/velocardiofacial syndrome include palatal clefting, velopharyngeal insufficiency, subtle dysmorphism, immunodeficiency, hypoparathyroidism, neurodevelopmental delay, and congenital heart disease typically involving the ventricular outflow tracts. Congenital heart defects occur in approximately three quarters of cases, with tetralogy of Fallot, interruption of the aortic arch, and common arterial trunk representing the most frequently associated cardiac abnormalities.Reference McDonald-McGinn, Kirschner and Goldmuntz 2 By comparison, there are relatively few reports of duplication of the chromosome 22q11 region, which in theory should occur with equal incidence as a deletion in the same region, as a result of reciprocal events caused by low copy repeat-mediated rearrangements.Reference Yobb, Somerville and Willatt 3 It is possible, however, that this syndrome is underdiagnosed as its wide spectrum of clinical findings makes it difficult to recognise clinically. Diagnosis is often made by default, usually when investigating the possibility of deletion 22q11.2 syndrome in an infant with a congenital cardiac defect. The duplication is often inherited from a normal or mildly affected parent, and thus the interpretation of this finding in an affected infant/individual can be difficult. If one parent has the duplication, then their recurrence risk is 50%, and the affected child also has a 50% risk of passing on the duplication to his/her children. If the duplication is a de novo event, the recurrence risk for parents is low.
Edelmann et alReference Edelmann, Pandita and Spiteri 4 first reported on duplication 22q11.2 in a 4-year-old girl. Ensenauer et alReference Ensenauer, Adeyinka and Flynn 5 helped define the phenotype of this new syndrome in their report on 13 individuals with microduplication 22q11.2. The clinical findings in patients with this syndrome are extremely variable, ranging from normal phenotype to mild learning difficulties, cognitive and behavioural problems, dysmorphic features, hearing loss, heart defects, urogenital abnormalities, velopharyngeal insufficiency, and cleft palate: a phenotype not dissimilar to that of DiGeorge/velocardiofacial syndrome. The TBX1 gene, located in band 22q11.21, has been shown to be the major disease gene responsible for DiGeorge/velocardiofacial syndrome. Overexpression of TBX1, as in our patient and others with duplication 22q11.21, has been observed to have the same clinical effect as loss-of-function mutations or deletions of TBX1. This would help to explain the somewhat similar phenotype in both deletion and duplication of this region of 22q and suggests that TBX1 is the major disease gene in this syndrome also.Reference Portnoï 6
Congenital cardiac defects have been reported in association with duplication 22q11.2 syndrome and include tetralogy of Fallot, hypoplastic left heart syndrome with interruption of the aortic arch, lethal congenital nonconotruncal complex heart defects, double-outlet right ventricle with transposed great arteries, total anomalous pulmonary venous return, d-transposition of the great arteries, ventricular septal defect, tricuspid atresia, and patent ductus arteriosus.Reference Laitenberger, Donner, Gebauer and Hoehn 7 – Reference Weisfeld-Adams, Edelmann, Gadi and Mehta 10 Our report describes the first case of hypoplastic left heart syndrome with hypoplastic pulmonary arteries in association with chromosome 22q11.21 microduplication syndrome in the absence of an interruption of the aortic arch. Successful palliation in our patient has allowed for growth of the pulmonary arteries facilitating progression to a successful extracardiac Fontan procedure.
In conclusion, array comparative genomic hybridisation is the investigation of choice in a patient with suspected microdeletion or microduplication of 22q11.2 as this abnormality cannot be detected on routine karyotype analysis. Array comparative genomic hybridisation has the potential to increase the diagnosis of genetic defects associated with hypoplastic left heart syndrome and other syndromes that include congenital cardiac lesions. Gene duplications, as well as gene deletions, may give rise to cardiac defects as demonstrated in this report.
Acknowledgement
The authors are grateful to Mr Andrew Pendred and Ms Deirdre Devlin for their assistance in preparation of figures for this manuscript.
Financial Support
This research received no specific grant from any funding agency, commercial or not-for-profit sectors.
Conflicts of Interest
None.


