Marfan syndrome is an autosomal dominant disorder with complete penetrance, characterised by mutations in the fibrillin gene (FBN-1), affecting the formation of connective tissue.Reference Judge and Dietz 1 The neonatal form is the most rare – prevalence of 1 to 6 out of 10,000 individuals – and it is a particularly severe form of the disease, limiting prognosis.Reference Hsien-Yu, Wan-Shiung and Te-Jen 2 Early mortality from neonatal Marfan syndrome is mainly due to complications in the cardiovascular system – aortic dissection, severe mitral regurgitation. The longest follow-up described in the literature, to our knowledge, is a child who survived into the fourth year of life.Reference ter Heide, Schrander-Stumpel, Pals and Delhaas 3
The following is the report of a child with neonatal Marfan syndrome, currently 11 years of age, who had undergone the Bentall-De Bono procedure at 2 years of age and thereafter two mitral valve replacements, with satisfactory late clinical evolution so far.
Case study
This is a male child, born through caesarean delivery at term, weighing 3630 grams and having a length of 51 centimetres, after an uneventful pregnancy and no relevant family history. In the hospital nursery, a suspicion of neonatal Marfan syndrome was raised. She had no clinical heart failure, but the echocardiogram in the first week of life demonstrated dilatation of the aortic root without significant valve regurgitation.
The child was seen initially at our institution at 5 months of age. On examination she showed skeletal signs of Marfan syndrome with long limbs and hyperflexibility, arachnodactyly, pectus carinatum, scoliosis, foot varus and facies with low-set ears and eyes, and an aged appearance. She was eupneic, ruddy, hydrated, and made no effort at being breast fed. She showed a heart rate of 150 beats per minute, blood pressure of 79/50 millimetres of mercury, and tachycardic heart sounds without murmurs. There were no signs of heart failure. An echocardiogram showed moderate annulus-aortic ectasia and mild aortic reflux, mitral valve prolapse with minimal regurgitation, and tricuspid valve prolapse without regurgitation (Fig 1). The electrocardiogram and chest X-rays had no major changes.

Figure 1 Transthoracic echocardiogram, parasternal long axis view, revealing significant aortic root dilatation with eccentric mild aortic valve regurgitation (small arrow).
During follow-up, we observed rapid growth of the aortic root (5 millimetres per year) and worsening of the aortic valve regurgitation.
At 2 years of age, the dilation of the aortic root (31 millimetres) and aortic reflux reached a significant degree, leading to ventricular dilatation (35 millimetres; Fig 2) and onset of fatigue during exercise. The child underwent surgical correction of the annulus-aortic ectasia by the technique of Bentall-De Bono – Dacron valved tube with Saint Jude mechanical prosthesis number 22 and reimplantation of the coronary arteries, with good results. She was maintained on beta-blockers and acetylsalicylic acid 100 milligrams per day and is doing well. At the age of 4 years, she had a spontaneous rupture of chordae in the mitral valve, leading to severe regurgitation (Fig 3) and congestive heart failure. The child then successfully underwent mitral valve replacement by bioprosthesis. After 3 postoperative years, she developed mitral bioprosthetic dysfunction with stenosis secondary to calcification, congestive heart failure, and severe pulmonary hypertension, requiring replacement with a new mitral bioprosthesis.

Figure 2 Evolution of the diameters of the aortic root (Ao) and left ventricle (LVDD).

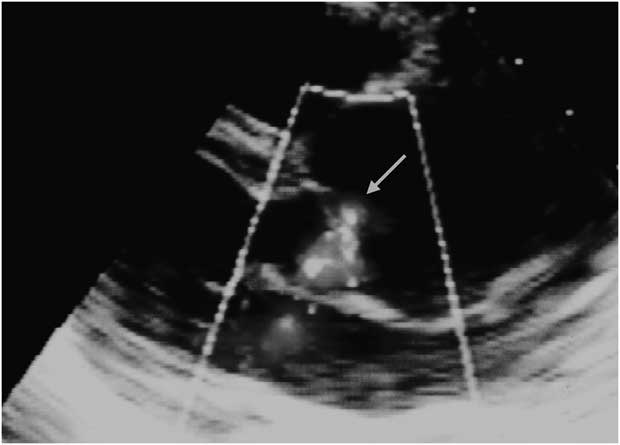
Figure 3 Echocardiographic long axis view showing a large flail as a result of mitral chordae rupture of the posterior leaflet (large arrow). A mechanical prosthesis is seen in aortic position (small arrow).
Currently, the child is 11 years old, weighs 26 kilograms, and is clinically stable. She has sinus rhythm, no signs of heart failure, and is on atenolol 25 milligrams per day and acetylsalicylic acid 100 milligrams per day. The echocardiogram demonstrates preservation of her left ventricular function, with normal functioning of the aortic and mitral prosthesis.
Discussion
This case, to our knowledge, is the first report on neonatal Marfan syndrome with favourable late outcome (9 years after first surgery) after three cardiac surgical interventions and treatment with a beta-blocker.
The neonatal Marfan syndrome, like the late adult form, is a connective tissue disorder that primarily affects the musculoskeletal, ocular, and cardiovascular systems. It has the peculiarity of rapid and progressive cardiovascular complications and early death.Reference Judge and Dietz 1 Its main differences from the classic Marfan syndrome are as follows: it always arises from new mutations (no family history) and presents with joint contracture, scoliosis, and joint dislocations. Multivalvular heart disease is the primary cardiac manifestation, compared with the involvement of the aorta and the aortic valve in adults, with prevalence of mitral, tricuspid, and pulmonary regurgitations. In addition, children with neonatal Marfan syndrome have a tendency to suffer from heart failure, leading to early death in most cases within the first year of life. The average life span of no more than 2 years is due to the severity of cardiovascular complications.Reference ter Heide, Schrander-Stumpel, Pals and Delhaas 3
This case, in line with the above description, has no family history and presented with ascending aorta aneurysm, multivalvular dysfunction, and early congestive heart failure. Fortunately, the clinical course was modified by therapeutic interventions and clinical surgery. As it has early clinical manifestations, neonatal Marfan syndrome is still a challenge, and the key issue for its effective treatment depends upon early diagnosis. However, in most patients, the syndrome is detected only when a serious complication has occurred with resulting poor prognosis.
In this case, early diagnosis was possible because of exuberant and peculiar phenotypic neonatal manifestations in the child, leading to suspicion by the neonatologist, confirmation by the geneticist, and early referral to a specialised service. The participation of the neonatologist, alert to the possible diagnosis of neonatal Marfan syndrome, was crucial in this case, allowing proper and timely medical and surgical treatment.
Initially, we decided on medical therapy using a beta-blockerReference Shores, Berger, Murphy and Pyeritz 4 as there was no sign of a significant aortic lesion or any valve dysfunction. The replacement of the aortic root and aortic valve was indicated when the children presented with signs of fatigue and left ventricular dilation. She reacted well to the de Bonno procedure without significant gradient through the Saint Jude prosthesis and we kept her only on aspirinReference Nishimura, Carabello and Faxon 5 without significant thromboembolic episodes or prosthetic dysfunction. She also had two mitral bioprostheses inserted, the last one 4 years ago and without any signs of dysfunction until now.
Currently, the child continues with her periodic evaluations at our institution, presenting herself with good cardiovascular stability and no signs of prosthetic dysfunction.