The history of cavopulmonary surgery
There is a wonderful and rich history about the origins of the classic cavopulmonary shunt which can be found in a number of recent and more remote references.4–9 Widespread appreciation, and then clinical application, of the cavopulmonary shunt likely began with the publications of Glenn et al. in the early-to-mid 1950s, and then with his publication in the New England Journal of Medicine in 1958.10–13 Yet others from Italy, the Soviet Union, the United States, and other countries had even earlier, or coincident with Glenn, conceived of this shunt.4–9, 14 Over the next decade, the classic cavopulmonary shunt, soon referred to by many as the Glenn shunt, although some eschew this designation,10, 11 gained wide acceptance and integration into clinical practice. It is likely that it was Haller et al. at the Johns Hopkins Hospital who first reported the experimental surgical construction of the bidirectional cavopulmonary shunt in dogs.15, 16 According to Trusler in his William Glenn lecture to the American Heart Association in 1989,6 the first report in humans of construction of an end-to-side bi-directional cavopulmonary shunt was that given by Azzolina et al. in 1972, having been performed in their first patient in May, 1969.17 Abrams had, even earlier, performed an end-to-side bidirectional cavopulmonary shunt in 1967, although his experience was not documented until considerably later in the paper by Salmon et al.18 thus overlapping with early descriptions of the Fontan procedure.19 Hopkins et al. in 1985,20 and then others provided, a physiologic rationale for the use of this shunt, while Bridges and her colleagues in 1990 advocated the use of the bidirectional cavopulmonary anastomosis as interim palliation for the high-risk Fontan candidate.21 Both of these observations set the stage for wide acceptance, and integration into clinical practice, of the bidirectional cavopulmonary shunt, an operation subsequently used widely in the staged palliation of the functionally univentricular heart.22
Late deterioration of the cavopulmonary shunt and pulmonary arteriovenous fistulas
Within a decade and a half of the appearance of Glenn's publication in the New England Journal of Medicine, other publications addressing reasons for late deterioration began appearing in the literature.22–24 These have been discussed and illustrated elsewhere25, 26 (Table 1).
Table 1.
Reasons for late deterioration of the cavopulmonary shunt.
Amongst a number of reasons for late failure of the classical Glenn shunt, Mathur and Glenn, in 1973, were the first to document the acquisition of pulmonary arteriovenous malformations as one phenomenon responsible for late clinical deterioration.27 Some erroneously attribute to McFaul et al. the earliest recognition of this complication of creation of the cavopulmonary shunt, but this is clearly incorrect.28 In the discussion given by Mathur and Glenn of a patient who became severely hypoxic after construction of a classic cavo-pulmonary shunt, they state: “This suggests, together with the angiographic findings, that most of the caval blood entering the right lung passes into the lower lobe and through widely dilated arteriovenous connections into the pulmonary vein without passing through the capillary circulation.”27 They did not, however, seemingly appreciate the egregious nature of this complication. Four years later, in 1977, McFaul et al. fully documented this complication in four patients following construction of a classical cavopulmonary shunt to palliate cyanotic congenital heart disease.28 They concluded on the basis of their observations: “Since this acquired AV malformation destroys the gas exchange capabilities in the right lung, we have been led to the conclusion that this procedure is presently the least attractive palliative shunt available for children with cyanotic congenital heart disease.”28 We have observed pulmonary arteriovenous fistulas to develop in the left lung following the Laks modification of the Fontan procedure when the superior caval vein is connected to the left lung, and inferior caval venous blood is diverted to the right lung in the setting of an adjustable atrial septal defect22, 29 (Table 2).
Table 2.
The cavopulmonary shunt and the development of pulmonary arteriovenous malformations: a chronology.
Pulmonary arteriovenous fistulas: issues about incidence and methodology for detection
There is no doubt that the classic cavopulmonary shunt provided excellent palliation for some patients with cyanotic congenital heart disease and reduced pulmonary blood flow.6, 22, 30–34 Indeed, as articulated by Castaneda, Trusler and Robicsek amongst others, the cavopulmonary shunt evolved in clinical practice to complete right heart bypass, the Fontan operation, and then to its many surgical variations.4–7, 19 Many of these surgical variations designed to be more energy-efficient began in 1988 with the experience of de Leval et al. with their total cavopulmonary connection.35 In the years following the publication of McFaul et al.,28 a number of reports further defined this complication, addressing the incidence of this development following classic cavopulmonary shunting, methodologies for their detection, and possible aetiologies.24, 26, 27, 30–34 Kopf et al. at Yale opined that the incidence of the development of pulmonary arteriovenous malformations at 10 years is 10%.32 Cloutier et al. in 1985, suggested that at least one-quarter of patients following the classic Glenn procedure, using the methodologies available at that time, develop pulmonary arteriovenous fistulas.24 It is likely that the incidence of acquiring pulmonary arteriovenous malformations after the classic Glenn shunt is probably even higher than this. Clinical observations, furthermore, suggest this process is not static. In the setting of the bidirectional cavopulmonary shunt, this complication is found bilaterally. It may, in fact, be a universal consequence of such interventions, although in many patients this is subclinical.36–39 Coincident with, and indeed paralleling the many clinical observations about the development of the pulmonary arteriovenous malformations occurring in the setting of cavopulmonary surgery, was the evolution of methodologies designed for their detection or recognition. This evolution began with standard chest radiography, followed by angiographic imaging (Figs 1 and 2), then by radionuclide technology (Fig. 3), and more recently by bubble/contrast echocardiography (Fig. 4) and computerized tomography, amongst other techniques.24, 25, 36–39
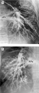
Figure 1.
Pulmonary arteriovenous malformation in two different patients. (a) Left pulmonary arteriogram (LPA) from a patient with bidirectional cavopulmonary anastomosis shows early and dense opacification of the left pulmonary vein (LPV). There are small grape-like vascular pools in the left mid-lung zone. (b) Right pulmonary arteriogram (RPA) from a patient who developed pulmonary arteriovenous malformation after the bidirectional cavopulmonary anastomosis shows persistent arteriovenous malformation after the modificed Fontan operation. Note numerous fistulous communications between the pulmonary arterial and venous branches in the peripheral lung and early opacification of the right pulmonary vein (RPV).
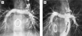
Figure 2.
Improvement of pulmonary arteriovenous malformation after Fontan operation. (a) Subtle vascular change is seen in the left lung in this patient with bidirectional cavopulmonary anastomosis. (b) After the modified Fontan operation with hepatic vein inclusion the vascular change in the left lung has largely disappeared. LSVC: left superior vena cava.
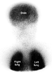
Figure 3.
Radioisotope pulmonary perfusion scan with injection of radionuclide-labelled microspheres through an arm vein in a patient with bidirectional cavopulmonary anastomosis shows radioisotope uptake in the brain as well as both lungs, suggesting pulmonary arteriovenous shunt. However, a similar result can be seen when there is collateral venous drainage of the superior caval venous compartment into the inferior caval venous system. (Courtesy of Dr Soo Jin Kim, The Sejong Heart Institute, Korea.)
Figure 4.
Bubble contrast echocardiogram in a patient with bidirectional cavopulmonary anastomosis shows echogenic bubbles in the left atrium (LA) and left ventricle (LV), suggesting pulmonary arteriovenous shunt. The patient had tricuspid atresia.
Bubble/contrast echocardiography is very sensitive for the detection of pulmonary arteriovenous fistulas.36–39 Any number of possible aetiologies were offered to explain the development of such pulmonary arteriovenous malformations after the Glenn procedure. The most popular amongst these for a number of years were absence of pulsatile pulmonary blood flow to the right lung, and dependent flow to the right lower lobe, reflected in a decreased ratio of perfusion between the upper and lower lobes.24 This latter finding was thoroughly documented in 1969 by Samanek et al.,40 using a radioisotope technique, and then by others. Data about the incidence of the development of the malformations following the classical Glenn, bidirectional cavopulmonary anastomosis, Fontan operation or Kawashima-type of total cavopulmonary connection is conflicting. Some of the disparity in frequency of this complication likely reflects the era in which the determination was made and, in turn, the methodologies employed. Feinstein has shown that contrast echo is very sensitive in this determination, much more so than angiography, and often positive despite normal pulmonary venous saturation.39 Thus, some degree of pulmonary arteriovenous shunting may be subclinical. Other factors perhaps contributing to differing frequencies may be the age of the patient when surveyed for this complication.36 Mahle et al., using pulmonary angiography, found that only 10 of 372 patients developed pulmonary arteriovenous malformations following a bidirectional cavopulmonary shunt.41 These 10 patients, with a variety of complex congenital cardiac malformations, had undergone the bidirectional cavopulmonary shunt at a mean age of 8.8 months, and the mean interval from bidirectional cavopulmonary shunt to catheterization was 10.9 months. Again, this study using angiography as the arbiter of pulmonary arteriovenous malformations will certainly underestimate the true frequency of this complication. It is not surprising, therefore, that these observations are at odds with those of Kim et al.37 Using a variety of methodologies, they reported that most patients with a bidirectional cavopulmonary shunt have subclinical evidence of right-to-left intrapulmonary shunting.37 Similar observations were published by Vettukattil et al.36 who, using a radionuclide determination, found that virtually all patients develop some degree of intrapulmonary right-to-left shunting after construction of a bidirectional cavopulmonary shunt.36 Bernstein et al., using contrast echocardiography, reported that three-fifths of children who had undergone a cavopulmonary shunt, all, with the exception of one patient, having undergone a bidirectional shunt, developed pulmonary arteriovenous fistulas.42
With the introduction of the Fontan principle in 1971, the classic Glenn cavopulmonary shunt came to be largely abandoned by the end of the 1970s as a form of functional univentricular palliation.43 Furthermore, since pulmonary arteriovenous malformations were considered uncommon in the classic Fontan operation despite absence of pulsatile blood flow into the lungs, this aetiology was largely discarded.24 The exact incidence of the development of clinically important pulmonary arteriovenous fistulas following Fontan surgery is unclear.24, 44 Again, distinctions should probably be made between those patients who develop clinically important fistulas, and those who demonstrate only a positive contrast echo study, although conceivably the latter could evolve into the former.
Intersection with the hepatopulmonary syndrome
Within a decade of the speculations of Cloutier et al., in 1985, as to the possible aetiology for the development of pulmonary arteriovenous fistulas following a classic cavopulmonary shunt,24 another tantalizing explanation was offered for that aetiology. Perhaps one should not be surprised that the liver was considered to play a role in their development45–55 (Table 3).
Table 3.
The liver and the development of pulmonary arteriovenous fistulas: a chronology.
By the mid 1990s, a number of observations dating back at least 10–15 years, and converging or overlapping with diverse clinical perspectives, much like Venn diagrams, led some to suggest that the liver might have a role in the development of acquired pulmonary arteriovenous malformations subsequent to cavopulmonary shunting.45–55 Advanced liver disease, and portal hypertension, produces a number of intrathoracic complications that involve the pleural space, the pulmonary parenchyma, and the pulmonary circulation. It is well known, that amongst patients with cirrhosis, up to one-third or more will develop arterial hypoxaemia secondary to intrapulmonary vascular dilations, leading to the designation of hepatopulmonary syndrome.46–48, 52–60 Intrapulmonary right-to-left shunting can also be documented in some patients with acute hepatic dysfunction, and others with non-cirrhotic portal hypertension.55 Historically, Fluckiger first reported the association of cyanosis with cirrhosis in 1884.56 Snell, in 1935, then described the finding of reduced oxygen saturation in a patient with cirrhosis.57 In 1956, Rydell and Hoffbauer reported multiple pulmonary arteriovenous fistulas in a patient with juvenile cirrhosis.58 It was Berthelot et al. who then documented the arterial changes in the lungs of cirrhotic patients.59 They characterized the intrapulmonary vascular abnormalities to consist of parenchymal and pleural capillary dilations and arteriovenous malformations.59 These patients with chronic and severe liver disease also have pulsatile pulmonary blood flow. Kennedy and Knudson coined the term “hepatopulmonary syndrome” in 1977 when discussing a cirrhotic patient with exercise-aggravated hypoxaemia and orthodeoxia.60 The observations of pulmonary arteriovenous malformations in the adult with acute or chronic liver disease have now been extended as well to children with cirrhosis. Many of these children are those with the complex of biliary atresia, polysplenia, and interruption of the inferior caval vein,46–48, 50, 51 amongst other children with chronic hepatic dysfunction or portal hypertension from other aetiologies. From clinical observations of both the children and adults with the hepatopulmonary syndrome, hepatic transplantation seemingly allows regression of pulmonary arteriovenous shunting, presumably by making available a specific hepatic factor that mediates or promotes advantageous remodelling of the pulmonary vascular bed.47, 48 The specific mechanisms responsible for this will be discussed later in this review. A similar pathophysiology may be seen in those patients with diffuse acquired pulmonary arteriovenous fistulas secondary to a patent venous duct, a malformation which results in a congenital portosystemic shunt.61–63 Ligation of the patent venous duct has been followed by resolution of the pulmonary arteriovenous fistulas. Banding of the patent venous duct has also proved beneficial in this regard in another patient.61–63
Pulmonary arteriovenous fistulas, the Glenn shunt, the Fontan and total cavopulmonary connection: the role of hepatic venous exclusion
One of the earliest, if not the earliest, report of development of pulmonary arteriovenous malformations after the Fontan procedure was that of Moore et al. in 1989,64 some 18 years after the benchmark publication from Fontan and Baudet.19 They described two patients. In their first patient, who had left isomerism, a Fontan-type operation was carried out at 5 years of age where, as part of the operation, the hepatic veins were connected only to the left lung. Progressive clinical deterioration began at 8 years of age, and by 10 years of age, pulmonary arteriovenous fistulas were demonstrated by angiography, but only in the right lung. Their second patient, also with left isomerism, underwent a total cavopulmonary connection, with the hepatic veins continuing to drain into the systemic venous atrium. Pulmonary angiography, and saline contrast echocardiography, demonstrated bilateral pulmonary arteriovenous fistulas.64 In neither of these 2 patients, however, was a hepatic factor, or exclusion of the hepatic veins from the pulmonary arterial circulation, considered as causal. Indeed, the aetiology of the pulmonary arteriovenous fistulas in these two patients was not discussed, although they did cite the publication of Cloutier et al. as documenting that patients are a group “at risk” for this development after the Fontan procedure.24 In 1993, Jonas published an invited letter concerning the importance of pulsatile flow when systemic venous return is connected directly to the pulmonary arteries.65 In the body of this letter, he states: “Perhaps the absence of some important interaction between a hepatic factor and lung blood vessels induces formation of arteriovenous malformations.” It was not until 1995, with the observations of Srivastava et al. from the Children's Hospital in Boston, that specific attention was focused on some putative hepatic factor that might have a role in the genesis of the pulmonary arteriovenous malformations in patients with congenital cardiac malformations.66 It is interesting that the letter by Jonas, with its speculation about a hepatic factor, was not cited by Srivastava et al.66 The group from Boston, nonetheless, was aware of the relationship between cirrhosis and pulmonary arteriovenous fistulae. They recognized that a proportion of patients who had undergone a classical Glenn shunt developed pulmonary arteriovenous fistulas in the shunted lung, this being excluded from receiving hepatic venous blood. They also appreciated that when, in the course of surgical treatment of some patients with complex congenital cardiac disease, hepatic venous blood was excluded from the pulmonary circulation, such patients were likely to develop pulmonary arteriovenous malformations. This group of patients were primarily, though not exclusively, those with polysplenia, left isomerism and interruption of the inferior caval vein who had been treated with a Kawashima-type total cavopulmonary connection.67, 68 The surgical concept of excluding the hepatic veins from the pulmonary circulation was apparently first carried out by Kawashima et al. in 1978, in patients with left isomerism, azygos continuation of the inferior caval vein, and complex congenital cardiac disease.67 They published their clinical experience with this modification of a total cavopulmonary connection in 1984, with further experience and follow-up over the next decade.68–70 In this modification of the total cavopulmonary connection, after completion of the operation, only the hepatic veins continue to drain or empty into the systemic venous atrium, thus being effectively excluded from the pulmonary circulation. The remainder of the systemic venous circulation is conveyed to the lungs by means of a connection using the azygos connection with or without a unilateral cavopulmonary connection should the brachiocephalic vein be absent or severely hypoplastic. The Kawashima operation was initially thought to afford or convey a circulatory advantage, as the hepatic venous flow would contribute to the systemic output, especially if the haemodynamics were not favourable after the connection, functioning in the same way as fenestration.68–70 Furthermore, by leaving the hepatic veins connected to the systemic venous atrium, the procedure avoided the need for a complex intra-atrial baffle and the potential for late rhythmic or mechanical problems related to its placement.68–71 Following the Kawashima total cavopulmonary connection, the saturations of oxygen in these patients are not normal, reflecting the contribution of unoxygenated hepatic venous flow to the systemic circulation. Clinical observations in these patients revealed that some would, over the years, become progressively hypoxemic due to the development of pulmonary arteriovenous fistulas.66–71 Kawashima et al. reported half of their patients with the total cavopulmonary connection experiencing a 10% or greater decline in saturation of oxygen secondary to the development of such arteriovenous malformations.68 When considering the evolution of the Kawashima-like operations, it should be noted that Kreitmann et al. performed a similar operation as illustrated in their case report of 1982,72 albeit that the Japanese experience had begun in 1978, giving Kawashima primacy to this novel approach to the total cavopulmonary connection.68
After the sentinel observations of Srivastava et al., a large number of publications supported the observations that hepatic venous exclusion from the pulmonary circulation was causal or contributory to the formation of pulmonary arteriovenous malformations.37–39, 41, 42, 73, 74 Yet there are some patients with polysplenia and interruption of the inferior caval vein with apparently normal hepatic function who have not undergone cavopulmonary surgery but who still develop pulmonary arteriovenous malformations.66, 75–77 Interestingly pulmonary arteriovenous fistulas have not yet been identified in unoperated patients with right isomerism, although this group must be most uncommon.78 The patient reported by Papagiannis et al., for example, was found to have hypoplasia of the intrahepatic portal venous branches and a portal-systemic shunt.79 The consequence of this was that mesenteric venous return bypassed the liver, and was thus conducted into the heart and lungs without metabolic additions or reductions.79 Such patients are similar to those with Abernethy syndrome, who also develop progressive hypoxaemia.80, 81 It is unclear, therefore, whether the development of pulmonary arteriovenous malformations in patients with hepatic venous exclusion is an “all-or-none” phenomenon.71
It is apparent that it takes time for the pulmonary arteriovenous malformations that develop in the setting of cavopulmonary surgery to be recognized clinically, often years, although this is not invariably so.6, 22, 24, 28, 31–33 Data published by Bernstein et al. also suggests that patients with heterotaxy may be at an increased risk for developing pulmonary arteriovenous fistulas after cavopulmonary shunting.42 Similar conclusions were reached by McElhinney et al., who recommend that such patients be provided with an additional source of flow of blood to the lungs.82 Some, nonetheless, have reported very rapid development of pulmonary arteriovenous malformations following a bidirectional cavopulmonary shunt.83 This suggests that recruitment and dilation of pre-existing vascular channels was probably responsible for their development, rather than true angiogenesis, which would take time.84 In this regard, we have demonstrated that systemic venous collaterals may be unmasked with great rapidity, following a bidirectional cavopulmonary shunt, again presumably from dilation of pre-existing vascular channels.85 Such pre-existing vascular channels were demonstrated nearly 40 years ago by Anabtawi et al., although much of the data presented in their paper was derivative.86 Very rarely, intrapulmonary arteriovenous shunting can be seen rapidly after biventricular surgical repair of pulmonary atresia and ventricular septal defect, again likely reflecting recruitment and dilation of pre-existing vascular channels rather than from angiogenesis.87
Kawashima et al. speculate that pulmonary arteriovenous malformations occur only in a minority of patients following a total cavopulmonary shunt operation, presumably with hepatic venous exclusion, and seldom occur in older patients.71 Kawashima et al. also suggest that older patients develop a collateral circulation that delivers this putative hepatic factor to the lungs, thus preventing disadvantageous lung vascular remodelling.71 Subsequent to the publication of Srivastava et al., who suggested that redirection of hepatic venous blood to the pulmonary bed might reverse the development of pulmonary arteriovenous malformations, a number of groups employed this manoeuver, with varying degrees of success. Some reported that redirection of hepatic venous blood to the pulmonary circulation was indeed curative.66, 71, 73, 74, 88 Even when hepatic venous blood is conveyed to the lungs after an earlier Kawashima-type operation, maldistribution of hepatic venous blood could still result in pulmonary arteriovenous malformations in the disadvantaged lung, as reported by Uemura et al.89 This may necessitate surgical redistribution of hepatic venous blood flow in a more equitable fashion, as shown by Uemura and Justino and their respective colleagues.89, 90 A number of specific surgical manoeuvers have been introduced to convey hepatic venous blood to the lungs, depending on the type of surgical cavopulmonary connection, and relative topography of the pulmonary arteries and hepatic veins.73, 74, 91, 92 Reconstitution of pulmonary arterial continuity after a Fontan procedure in a patient with a previous Glenn shunt has also been reported to lead to complete resolution of pulmonary arteriovenous fistulas. Cardiac transplantation has also led to regression of pulmonary arteriovenous malformations, again presumably by redirecting hepatic venous effluent to the pulmonary circulation.93
Heart malformations with congenital hepatic venous exclusion
Evidence from other forms of congenital cardiac disease also lent support to hepatic venous exclusion as a likely aetiology for the development of pulmonary arteriovenous malformations. Lee et al. reported a cyanotic boy with pulmonary arteriovenous malformations who was found to have drainage of the hepatic veins and coronary sinus to the left atrium as the only cardiac anomalies, this resulting in the hepatic venous effluent being excluded from the pulmonary circulation.94 This boy was treated by diversion of the hepatic flow from the left atrium to the right atrium using autologous pericardium.94 By 5 months after the operation, the patient was no longer clinically cyanosed, and the saturation of oxygen, which had been 76% preoperatively, was now 100%. One year postoperatively, a bubble echocardiogram no longer showed abnormal cavitations in the left heart, consistent with complete regression of the pulmonary arteriovenous malformations. Another patient with complex totally anomalous systemic venous return, including connection of the hepatic veins to the left atrium, became increasingly cyanotic after the systemic venous return was effectively connected to the right heart, along with closure of a defect in the oval fossa, leaving the hepatic venous connected to the pulmonary venous atrium. The important pulmonary arteriovenous malformations resolved after the hepatic veins were re-routed to the systemic venous circulation, and thus into the lungs95 (Table 4).
Table 4.
Conditions with hepatic venous exclusion.
“Mimics” of pulmonary arteriovenous fistulas
Pulmonary arteriovenous malformations are but one explanation, excluding pulmonary disease, for pulmonary venous and left atrial desaturation following cavopulmonary surgery. We have summarized elsewhere those diverse venous anomalies which may contribute to pulmonary venous desaturation.22, 26, 43 From the superior compartment, these include a number of venous connections originating from the superior caval vein, brachiocephalic vein or left phrenic vein amongst others, connecting either to one or more pulmonary veins or the pulmonary venous atrium.31, 85, 96–103 Reopening of a left superior caval vein connecting to the coronary sinus, or the so-called levoatrial cardinal vein, will have the same affect.26 At the cardiac level, a wholly or partially unroofed coronary sinus, and/or anomalous connections of one or more cardiac veins connecting to the pulmonary venous atrium, will contribute to systemic desaturation.104–108 These peculiar connections between the right atrium or coronary sinus with the left atrium have necessitated a better understanding of the normal venoatrial connections,109 and the nature of the myocardial connections between left atrial myocardium and the muscular walls of the coronary sinus.110
Partial hepatic venous exclusion was advocated to optimize postoperative Fontan haemodynamics.111 It soon became apparent that the excluded hepatic vein may also contribute to a postoperative intrahepatic venovenous shunt, resulting in right-to-left shunting.112–115 As a congenital anomaly, a hepatic vein may connect to the pulmonary venous atrium. If unrecognized prior to a total cavopulmonary connection, this can lead to important hypoxaemia, with massive intrahepatic venovenous shunting.116–121 Similarly, a hepatic vein may connect to the coronary sinus. Then, when the coronary sinus is positioned in the pulmonary venous atrium at the time of a total cavopulmonary connection, an important right-to-left shunt may also result. Connections between the hepatic and pulmonary veins have also been described with similar pathophysiology. Thus, these diverse systemic venous collateral channels, or venous anomalies at supracardiac, cardiac or infracardiac level, may contribute to important right-to-left shunting after construction of a total cavopulmonary connection. Appreciation of those diverse anatomic causes for left atrial or pulmonary venous desaturation should obviate a false-positive contrast echo study when performed from a peripheral vein rather than from the pulmonary arteries (Table 5).
Table 5.
“Mimics” of pulmonary arteriovenous fistulas.
Manoeuvers to treat acquired pulmonary arteriovenous malformations in patients with congenital cardiac malformations
Intervention for some of the late reasons for failure of a Glenn shunt has only been occasionally satisfactory.122, 123 Such interventions include those to close a residual superior communication between the caval vein and the right atrium, ligate an azygos vein, or interrupt systemic venous collaterals. The therapy to treat pulmonary arteriovenous malformations developing after the Glenn shunt has also evolved. Some of the earlier forms of therapy included right lower lobe lobectomy, or pneumonectomy, or balloon or coil embolotherapy.124 None of these were particularly attractive in those patients who developed pulmonary arteriovenous malformations following cavopulmonary shunting because of the tendency to recurrence and redistribution. The introduction of the bidirectional cavopulmonary shunt as interim palliation brought new issues into play. This manoeuver, initially advocated for so-called high-risk Fontan patients, was soon applied in many centres to most patients on the Fontan tract, although some did not feel such staging was necessary.21, 22, 125, 126 In some patients, the bidirectional cavopulmonary shunt served as the near final surgical option for patients considered at high risk for completion of the Fontan procedure.127 Once attention was focused on the concept of hepatic venous blood and its exclusion from the pulmonary vascular bed, any number of surgical manoeuvers were introduced to convey hepatic venous blood to the lungs.73, 74, 88–95 Some advocated also including additional sources of flow of blood to the lungs in those patients with a bidirectional cavopulmonary shunt. We have summarized elsewhere the considerable literature that has discussed the pros and cons of accessory pulmonary blood flow in the patient with a bidirectional cavopulmonary shunt.22 Whether this affords any protection against the development of pulmonary arteriovenous fistulas is unclear. Bernstein and his colleagues suggest that the provision of an additional source of pulsatile pulmonary blood flow has little protective effect.42 The creation of a fistula from the axillary artery to vein was designed to augment flow of blood to the lungs in a patient with a failing Glenn shunt.128, 129 In this regard, we, like others, have created such a fistula to provide such a source of accessory pulmonary blood flow when treating patients with pulmonary arteriovenous fistulas.130, 131 From our limited clinical experience with this manoeuvre, it is unclear whether the patients benefited from this approach.131 Once a total cavopulmonary connection had been constructed with hepatic venous exclusion, and then later appreciating that these patients were at risk for developing pulmonary arteriovenous fistulas, a number of procedures were employed to convey hepatic venous blood to the lungs. These included intra-atrial baffles to divert hepatic venous blood into the lungs following a Kawashima-type operation, connection of the hepatic veins to the azygos vein either directly or via a conduit, fenestration of the Kawashima circuit, and so on. In those patients with an unequal distribution of hepatic venous flow to the lung, surgical revision to provide better symmetry of hepatic venous flow has also been performed.73, 74, 88–95
The biology of pulmonary arteriovenous fistulas after cavopulmonary surgery
Structure of pulmonary arteriovenous fistulas
There is some information on the architecture of the pulmonary arteriovenous fistula.86, 132 Considerable information has been assimilated from the ongoing clinical experience in the evaluation of patients with hereditary haemorrhagic telangiectasia, acknowledging that, in many of these patients, the pulmonary vascular abnormalities are larger, sac-like, and amenable to embolotherapy.133 Yet still the documented histopathology of pulmonary arteriovenous malformations is sparse. For example, in Spencer's exhaustive 1996 5th edition of Pathology of the Lung, less than one full page is devoted to pulmonary arteriovenous fistulas, and there is no mention of their development after cavopulmonary surgery.132 Until recently, there has also been a paucity of information about the structure of these malformations in children with congenital cardiac malformations following cavopulmonary surgery (Fig. 5).
Figure 5.
Pulmonary arteriovenous malformations in a patient dying after a Fontan operation. This 4-year-old child was born with pulmonary stenosis and multiple ventricular septal defects and a functional “single” ventricle). Had undergone pulmonary artery banding and then at 2 years of age had bilateral, bidirectional Glenn shunts. Before the Fontan operation he was found to have mild bilateral pulmonary arteriovenous fistulae, dying in the immediate post-Fontan time period. (a) This high power photomicrograph shows a dilated vein with associated dilated, thin wall tortuous vascular channels within the interlobar septae of the lung. (b) This low power photomicrograph of lung tissue from the same patient as (a) and shows medium sized arteries with their accompanying bronchi (arrows) and between them is a hugely dilated vein (arrowhead).
Whether or not the architecture at any level of these malformations differs between those occurring as isolated anomalies, those associated with the Weber–Osler–Rendu complex, those found after hepatic venous exclusion, or in those with cirrhosis is unclear. Duncan et al. have provided such information found at lung biopsy from two children with angiographically-proven pulmonary arteriovenous malformations following hepatic venous exclusion surgery.1 Histologic examination demonstrated large, dilated blood vessels, described as lakes, and clustered, smaller vessels or chains, in the pulmonary parenchyma. Density of the microvessel, reflecting an ongoing angiogenic stimulus, was significantly greater in these patients than in age-matched controls. immunohistochemistry demonstrated uniform staining for type IV collagen and alpha-smooth muscle actin, weak staining for the endothelial marker CD31, and negative staining for proliferating cell nuclear antigen. Electron microscopy revealed endothelial irregularity, a disorganized basement membrane, and increased numbers of collagen and actin filaments beneath the endothelium. The histologic correlate of this condition in children with congenital heart disease who have pulmonary arterial blood flow devoid of hepatic venous effluent appears to be a greatly increased number of thin-walled vessels.1 Immunohistochemistry suggests that the rate of cellular proliferation is not increased in these lesions.
Pulmonary arteriovenous fistulas and angiogensis
The past several decades have witnessed an explosion of clinical and experimental observations on the basic mechanisms responsible for angiogenesis and its regulation.134–138 Angiogenesis plays an important role in the pathology of cancer, ischaemic diseases, diabetic retinopathy, chronic inflammation, and hereditary haemorrhagic telangiectasia, among other conditions.135–139 One must distinguish between angiogenesis and vasculogenesis.139 Vasculogenesis is the formation of the first primitive vascular plexus. Angiogenesis, in contrast, is the formation of new vessels from pre-existing ones. Both processes are regulated by a delicate balance of pro- and anti-angiogenic factors.134–139 The process of angiogenesis also participates in the formation of pulmonary arteriovenous fistulas.1 One must acknowledge Folkman et al. for their many critical, and indeed benchmark, contributions to the field of angiogenesis, beginning with their observations of tumour angiogenesis factor, and then extending to intrinsic and extrinsic regulators and inhibitors of angiogenesis.135, 138, 140 We suggested some years ago, in the Mannheimer lecture, that the development of pulmonary arteriovenous malformations following the classic cavopulmonary shunt or bidirectional cavopulmonary shunt is a wonderful paradigm for bed-to-bench research.141 Such observations have now evolved, especially in the past decade, from those of a clinical nature or concern to resonate in more fundamental or basic investigations. This evolution was certainly stimulated in large part by the observations of Srivastava et al., with their appreciation of the development of pulmonary arteriovenous malformations secondary to hepatic venous exclusion,66 and also from observations in the liver-lung interface.45–55 Interest in the liver, and its role in the regulatory mechanisms of angiogenesis, seemed a natural evolution, paralleling those ongoing developments and investigations in vascular biology.142
The cascade of more fundamental studies began with detailed histologic observations of pulmonary arteriovenous malformations and those biological mechanisms responsible for remodeling the pulmonary vascular bed.1 The biology of the fistulas is indeed complex, and is intertwined with those diverse mechanisms that stimulate angiogenesis, or inhibit it. Lungs developing pulmonary arteriovenous fistulas after a cavopulmonary shunt express specific angiogenic factors.143–147 Starnes et al. have shown that specimens taken at lung biopsy from children after a cavopulmonary anastomosis demonstrate increased expression of vascular endothelial growth factor and its receptor.143, 144 These observations confirmed their earlier findings that blood vessels forming after a cavopulmonary anastomosis may have reduced intercellular junctions. They further suggest that vascular endothelial growth factor may be a mediator of angiogenesis in the lungs of children after the cavopulmonary anastomosis. They caution that other factors, such as vascular dilation and remodelling, may also be important to the formation of pulmonary arteriovenous malformations.143 Malhotra et al. have also discussed the possible role of angiotensin in the development of pulmonary arteriovenous fistulas.145, 146 Angiotensin, an endogenous bioactive peptide constituent of the renin–angiotensin system, is known to act as an inhibitory growth factor in vitro and in vivo.148–151 They found that, after a unilateral cavopulmonary shunt, the shunted lung had substantially lower levels of angiotensin-converting enzyme and angiotensin II compared with controls.145, 146 These changes were observed early after the cavopulmonary anastomosis, returning to normal levels 15 weeks after surgery. Malhotra et al. have subsequently published interesting findings about the role of oxidative stress in the development of pulmonary arteriovenous malformations after the cavopulmonary anastomosis.147 They studied gene expression in two groups of patients: those who underwent cavopulmonary shunting and those who had undergone banding of the pulmonary trunk. Both procedures resulted in an increase in the expression of the angiogenic gene, but only the cavopulmonary anastomosis induced the expression of endothelial stress-related genes. Vascular endothelial growth factor was upregulated several fold after both procedures. Only the cavopulmonary anastomosis, however, upregulated two stress-related genes, HO1 and GLUT1, respectively. Hypoxia-inducible factor was upregulated four-fold after the cavopulmonary anastomosis. Banding, in contrast, failed to induce the increased expression of any of these proteins. On the basis of this study, they conclude that reduced pulmonary blood flow induces a pulmonary angiogenic response but not an endothelial stress response. These results suggested that oxidative stress is more relevant to the formation of pulmonary arteriovenous malformations than angiogenic signalling alone, since banding did not result in pulmonary arteriovenous malformations. Oxidative stress of the pulmonary endothelium resulting from cavopulmonary anastomosis may predispose the affected vasculature to arteriovenous shunting. Mainwaring et al. have shown that patients with pleural effusions after the bidirectional cavopulmonary shunt demonstrate elevation in renin and angiotensin II on the fifth postoperative day as compared to patients who did not develop effusions.151 They conclude that patients who develop effusions following the bidirectional Glenn shunt have activation of their renin–angiotensin system. It should not be surprising, therefore, that inhibitors of angiotensin converting enzyme decrease the severity and duration of pleural effusions following a bidirectional cavopulmonary anastomosis. Hypoxia is a potent stimulator of angiogenesis, presumably on the basis of the upregulation of vascular endothelial growth factor.152–156 The fact that most patients with hypoxia secondary to congenital heart malformations do not develop pulmonary arteriovenous fistulas underscores the mechanistic complexity of disordered or upregulated angiogenesis. In order to define more precisely those biological factors contributing to the development of pulmonary arteriovenous malformations after the Glenn shunt, Starnes et al. have now produced a rat model of these vascular abnormalities.157 This model should prove helpful in defining the putative hepatic factor, and those other angiogenic stimulating and inhibiting factors participating in the formation of pulmonary arteriovenous fistulas after cavopulmonary shunting.
The liver and angiogenesis
A number of clinical and pathological observations support the view that the liver and portal circulation regulate in some fashion certain aspects of the pulmonary circulation.45–55, 61–63, 73–84, 94, 95 Marshall et al., in the perspective of cavopulmonary surgery, stated in 1997 that the liver is actively involved in the maintenance of normal pulmonary vascular integrity.1 They stated that pulmonary arteriovenous malformations developing after a cavopulmonary shunt represent a form of abnormal angiogenesis under hepatic control or regulation.1 The corollary to these observations is to identify those hepatic factors derived from, or modified by, the liver which influence the proliferation of pulmonary endothelium.142 Duncan and Desai have recently extended these observations, providing a thoughtful review of pulmonary arteriovenous fistulas developing after a cavopulmonary shunt.158 They have summarized the pertinent literature addressing the role of the liver in angiogenesis. Interestingly, the liver produces precursors of angiogenesis inhibitors which are transformed into endostatin and angiostatin.142 Plasminogen is the precursor of angiostatin, and collagen-XVIII is the precursor of endostatin. Hepatocytes are apparently the main source of these proteins, normally synthesizing and secreting them into plasma.135–138, 142, 159–163 Angiostatin, an inhibitor of angiogenesis, contains 3–4 kringle domains that are derived from proteolytic cleavage of plasminogen. The antiangiogenic effects of angiostatin occur, in part, from its inhibition of endothelial cell surface adenosine triphosphate synthase, integrin functions, and peri-cellular proteolysis. Angiostatin has structural similarities to hepatocyte growth factor, a promoter of angiogenesis, that induces proliferation and migration of both endothelial and smooth muscle cells via its cell surface receptor.164 The angiogenesis inhibitor endostatin is a 20 kDA C-terminal fragment of collagen-XVIII, a proteoglycan/collagen found in vessel walls and basement membranes. Endostatin inhibits endothelial cell migration and proliferation and induces their apoptosis.135–138, 161 Exclusion of these substances after a cavopulmonary anastomosis, or because of congenital hepatic venous exclusion, could result in angiogenesis, vascular proliferation, and eventuate in pulmonary arteriovenous fistulas.
The liver and pulmonary hypertension
A spectrum of structural and pathologic changes in the lung vasculature have been identified in patients with acute or chronic liver disease.165–172 The hepatopulmonary syndrome is part of the spectrum of pulmonary vascular disorders observed in some patients with advanced liver disease. We have devoted considerable discussion thus far in this review to pulmonary arteriovenous fistulas as a marker for the hepatopulmonary syndrome. In addition, pulmonary arterial hypertension is a well-known component of the liver-lung interface, and there is a substantial literature devoted to portopulmonary hypertension.165–172 Pulmonary arterial hypertension is observed in about 2% of patients with cirrhosis and portal hypertension.173 Because pulmonary hypertension can be seen in patients with non-cirrhotic portal hypertension, it is hypothesized that portal hypertension, and the derivative metabolic alterations, are likely the aggravating features.167, 170 In some patients, severe pulmonary arterial hypertension may be considered a contraindication to liver transplantation.52, 55, 165–172 This is not invariably so, and liver transplantation may lead to remodelling of the pulmonary vascular bed, with regression of the vascular changes.167–174 What is curious is how cirrhosis of the liver, and/or portal hypertension, can produce two very different vascular disorders of the pulmonary circulation, seemingly mutually exclusive, yet both reflecting some aspect of abnormal angiogenesis.167, 175 Pulmonary arterial hypertension, a disorder of angiogenesis, is typically characterized by endothelial and vascular smooth muscle proliferation within small pulmonary arteries.176–179 The structure of pulmonary arteriovenous malformations is distinctly different from pulmonary hypertensive disease, with loss of boundaries between arterioles and venules and a normal or low pulmonary vascular resistance in the latter.1, 132 Whether in some patients with the hepatopulmonary syndrome both vascular pathologies co-exist, but with only one dominating the clinical picture, is unclear. Jones et al. reported the clinical and laboratory investigations of a patient with co-existence of hepatopulmonary syndrome and portopulmonary hypertension.180 Their patient died, but a post-mortem examination was not performed.180 Krowka et al., with their considerable experience and many publications devoted to the interface between the liver and pulmonary vascular pathology,55, 165, 171, 175 have stated that he has not seen any autopsy specimen demonstrating true co-existence of diffuse vascular dilations and pulmonary arterial hypertension (M. Krowka, personal communication, January 22, 2004). Krowka has also written that the hepatopulmonary syndrome and portopulmonary hypertension represent two distinct pathological, clinical and diagnostic entities, each presenting their own unique challenges.181 Yet there are patients in whom pulmonary hypertension has developed following resolution of hepatopulmonary syndrome, suggesting to Mal et al. that these processes are not mutually exclusive.182 Other reports of similar evolution in pulmonary vascular pathologies following liver transplantation have also been published.183–185 Despite the differences between the hepatopulmonary syndrome and portopulmonary hypertension as articulated by Krowka, what potentially unites these disparate pathologies is the “biological umbrella” of disordered angiogenesis. One could make an analogy to genetic pleiotropy, where a single mutant gene results in the production of apparently unrelated multiple effects at the clinical or phenotypic level. Thus, one would wonder whether the designation of hepatopulmonary syndrome is still too limiting and non-specific. This designation could be expanded to embrace the entire spectrum of pulmonary vascular pathologies in those patients with acute or chronic liver disease or portal hypertension (Fig. 6).
Figure 6.
Schematic diagram shows in theory an expanded concept of the hepatopulmonary syndrome.
Hereditary haemorrhagic telangiectasia and pulmonary arteriovenous fistulas: relationship to pulmonary arteriovenous fistulas developing after cavopulmonary surgery
In the realm of speculation, what if any is the relationship between the pulmonary arteriovenous malformations occurring in the hereditary haemorrhagic telangiectasia complex and those fistulas developing after cavopulmonary surgery, or in the hepatopulmonary syndrome? Hereditary haemorrhagic telangiectasia, or Rendu–Osler–Weber disease, occurring in 1 to 5000 to 8000 people, is an example of a genetic disorder of angiogenesis in which a multi-systemic angiodysplasia results in diverse clinical findings, often associated with haemorrhage, adverse neurologic events, and so on.2, 186–190 So-called hereditary haemorrhagic telangiectasia type 1 is an autosomal dominant vascular dysplasia, caused by heterogeneous mutations in the endoglin gene on chromosome 9, and characterized by dilated vessels and arteriovenous malformations in the lungs, brain, liver and elsewhere.186–190, 191–197 Pulmonary arteriovenous malformations occur in up to one-quarter of patients with this type of hereditary haemorrhagic telangiectasia. The gene activin-receptor-like kinase 1 found on chromosome 12 is mutated in so-called “type 2” hereditary haemorrhagic telangiectasia. This form of hereditary telangiectasia is less frequent than the first type.186–189 Pulmonary arteriovenous malformations were found in only one-twentieth of those screened with the second type of hereditary haemorrhagic telangiectasia,196 a finding also reported by Abdalla et al.197 In the study reported by Kjeldsen et al., the prevalence of pulmonary arteriovenous malformations varied considerably depending on the specific mutation in the activin-receptor-like kinase 1 gene.198
Both endoglin and activin-receptor-like kinase 1 encode receptor members of the superfamily of beta-signalling transforming growth factor.186–189, 194, 195 Endoglin is a homodimeric membrane glycoprotein primarily expressed on endothelial cells. It is also an auxiliary receptor for the transforming growth factor family of cytokines, and is required for angiogenesis and cardiac development.194, 195 In association with transforming growth factor receptors I and II, it can bind transforming growth factor-beta1 and -beta3 and form a functional receptor complex. There is increasing evidence that endoglin can modulate the cellular response to transforming growth factor-beta, a factor implicated in formation of vascular lesions in human and experimental models.186–189 Endoglin is predominantly expressed on endothelium and is mutated in the first type of hereditary haemorrhagic telangiectasia. Several characteristic morphological and functional differences distinguish arteries from veins.191, 192 It was once thought that haemodynamic forces shaped these differences. Increasing evidence now suggests that morphogenetic programs have a central role in differentiation of blood vessels.192, 199–201 As a vascular dysplasia, hereditary haemorrhagic telangiectasia is characterized by the inappropriate fusion of arterioles with venules. The genes implicated in this disease, activin-receptor-like kinase 1 and endoglin, may be involved in defining the fundamental boundaries between arteries and veins.190, 194 Endoglin, activin-receptor-like kinase 1, and bone morphogenetic receptor type 2 participate in differentiation and proliferation of cells, and also apoptosis in embryonic and mature tissues.186–190, 194, 202 In a murine model of hereditary haemorrhagic telangiectasia, which is heterozygous for a targeted deletion in the endoglin gene, Torsney et al.203 observed intrinsic abnormalities in the vascular walls throughout the cutaneous vasculature. They found that postcapillary venules were dilated, and up to seven-tenths of the vascular wall lacked smooth muscle cells. The supporting layers of collagens and elastin were irregular and thin, adding to the fragility of these vessels. A variable haemorrhagic phenotype was observed in which local bleeding is associated not only with fragile vessels but also with regions of inflammation.203 Because of the considerable clinical heterogeneity in hereditary haemorrhagic telangiectasia, Berg et al.204 have asked whether pulmonary arteriovenous malformations are more common in families linked to endoglin? At least two different locuses have been shown for hereditary haemorrhagic telangiectasia. Mutations in endoglin have been found in some families, and the locus designated ORW1. In other families, this locus has been excluded. They confirmed that, in families linked to ORW1, pulmonary arteriovenous malformations are found in up to one-third of affected members, compared to less than one-twentieth in families in which this locus has been excluded.204
Even more intriguing is the relationship in some patients between hereditary haemorrhagic telangiectasia and primary pulmonary arterial hypertension.205 The pulmonary vascular pathology in these patients may exhibit both arteriovenous fistulas and the occlusive vascular pathology of primary pulmonary arterial hypertension.205 Among the earliest reports of these coexisiting pathologies was that of Sapru et al.206 They documented, in 1969, several patients with hereditary haemorrhagic telangiectasia and both pulmonary hypertension and pulmonary arteriovenous fistulas. A few years later, Trell et al.207 also reported this then uncommon combination of vascular pathologies in sisters. The clinical and pathological features of the pulmonary hypertension in those patients with hereditary haemorrhagic telangiectasia are indistinguishable from primary pulmonary arterial hypertension.205 The gene for primary pulmonary arterial hypertension is linked to the long arm of chromosome 2, at 2q 31-33.208–213 This has now been refined even further to heterozygous mutations within the gene bone morphogenetic protein receptor-2.208–212 Bone morphogenetic protein receptor-2 product is a receptor in the multifunctional transforming growth factor-beta signalling pathway. Both transforming growth factor-beta and bone morphogenetic proteins are necessary for differentiation, proliferation, and apoptosis of many types of cell, and have critical roles in embryogenesis.208–212, 214–216 Mutations in the genes for components of the transforming growth factor-beta signalling pathway underlie the inherited vascular disorder of hereditary haemorrhagic telangiectasia, and contribute to the pathogenesis of cancer and other diseases.208–212, 214–216 Trembath et al.205 have recently studied extensively the clinical and molecular genetic features of these patients with pulmonary hypertension in the setting of hereditary haemorrhagic telangiectasia. Amongst 10 patients with these unusual comorbid illneses, two also had pulmonary arteriovenous fistulas. Mutations were identified in the activin-receptor-like kinase 1 in the patients with both pulmonary hypertension and hereditary haemorrhagic telangiectasia.205 These mutations are associated with diverse effects, including the vascular dilations typical of hereditary haemorrhagic telangiectasia, and the occlusive arteriopathy characteristic of primary pulmonary hypertension. Harrison et al.,217 studying 11 probands with this combination of pathologies, have shown that the association of pulmonary arterial hypertension and hereditary haemorrhagic telangiectasia identifies an important complication appearing most commonly among subjects with defects in activin-receptor-like kinase 1 receptor signalling. Thus, like patients with liver disease and either pulmonary arteriovenous malformations or pulmonary hypertension, patients with hereditary haemorrhagic telangiectasia can exhibit one or the other pulmonary vascular pathologies. But, in those with hereditary haemorrhagic telangiectasia and pulmonary arterial hypertension, vascular telangiectasia has also been observed in the lungs.205–207, 217 The diverse pulmonary vascular pathology seen in patients with hereditary haemorrhagic telangiectasia is likely explained, at least in part, by the pleiotropic effects of the ligand for the transforming growth factor-beta receptor family, including transforming growth factor-beta, bone morphogenetic protein, and activin.205, 210, 217 This topic has been discussed in detail by Loscalzo.218 Thus, it is possible to define a “scrabble” of pulmonary arteriovenous malformations in and out of the setting of cavopulmonary surgery or the syndrome of hereditary haemorrhagic telangiectasia that intersect at many levels. The vascular biology of hepatic venous exclusion following cavopulmonary surgery culminates in pulmonary arteriovenous malformations, while the pulmonary vascular endpoints of the interface between lungs and liver are either pulmonary arteriovenous malformations or pulmonary vascular disease and pulmonary hypertension, apparently mutually exclusive, or nearly so. Activin-receptor-like kinase 1 and endoglin participate in the complex biology of both types of hereditary haemorrhagic telangiectasia, autosomal vascular dysplasias, with somewhat more than a quarter of the patients with the first type having pulmonary arteriovenous malformations. The biological and genetic intersection of those patients with hereditary haemorrhagic telangiectasia and primary pulmonary hypertension provides a unique model for the study of angiogenesis.219 Does the biology of endoglin also intersect with the liver and its putative hepatic factor, apparently excluded by those surgical manoeuvers preventing hepatic venous blood from reaching the lungs? Does the hepatic venous effluent differ between patients with and without hereditary haemorrhagic telangiectasia? The answers to these fundamental questions about the biology of pulmonary arteriovenous malformations will likely be unravelled in the early part of the 21st century as part of the Quixotic quest and desire further to understand the regulatory and modulating processes of angiogenesis.219 Until then, the specific pathogenesis of pulmonary arteriovenous fistulas will remain unknown. As a final thought, Robicsek4 more than two decades ago, wrote an epitaph for cavopulmonary surgery, a procedure which he had helped to pioneer. The epitaph was possibly premature, as even he concluded. The final irony is that, in the surgical palliation of the functionally univentricular heart, the bidirectional cavopulmonary shunt may in future be the sole manoeuvre not carried out in the catheter laboratory.220 Indeed, we must all await and anticipate the final soliloquy that will accompany the opening of Pandora's box of research, and other observations stemming from connecting a great vein to the pulmonary artery.
Conclusion
The following provides a summary of the previous discussion:
- Pulmonary arteriovenous malformations are a serious consequence of the classic cavopulmonary shunt, the bidirectional cavopulmonary shunt, and the variant of the total cavopulmonary connection introduced Kawashima.
- These pulmonary vascular abnormalities are far less common in patients who have undergone the Fontan procedure, where hepatic venous blood is incorporated into the pulmonary circulation.
- Pulmonary arteriovenous malformations occur in end-stage hepatic disease and may be reversed by transplantation of the liver.
- Hepatic venous exclusion is considered an aetiology for the development of pulmonary arteriovenous malformations after a cavopulmonary shunt.
- Hepatic venous inclusion into the pulmonary circulation usually leads to reversal of these pulmonary vascular malformations.
- These observations lend support to the role of a putative hepatic factor in the genesis of these arteriovenous malformations in patients with congenital cardiac malformations.
- Some degree of intrapulmonary right-to-left shunting may be a universal feature of the circulation subsequent to cavopulmonary shunting.
- The fundamental mechanisms regulating angiogenesis will intersect with those biologic processes that result in pulmonary arteriovenous malformations.
- The basic biological mechanisms responsible for these pulmonary arteriovenous malformations are being unravelled.
- A novel rat model involving the superior caval vein with the pulmonary arterial anastomosis will be valuable in the basic scientific quest.
- The vascular endpoints of the hepatopulmonary syndrome, namely either pulmonary arteriovenous malformations or pulmonary hypertension, are seemingly mutually exclusive.
- Any relationship between the pulmonary arteriovenous malformations occurring in hereditary haemorrhagic telangiectasia and those found after surgical cavopulmonary shunting or in the hepatopulmonary syndrome remains speculative.
- Fundamental knowledge about angiogenesis will likely be derived from understanding the molecular genetic features in patients with hereditary haemorrhagic telangiectasia and pulmonary hypertension.
- The bed-to-bench paradigm resonates in the cascade of knowledge accruing with regard to the course of pulmonary arteriovenous malformations occurring after creation of a cavopulmonary shunt.