Aortopulmonary window is a rare congenital heart lesion. It might be associated with other congenital heart diseases, as well as with anomalous origin of the coronary arteries. We are presenting a neonate, delivered prematurely, at 27 weeks of gestation, with a birth weight of 1 kg. Foetal echocardiography was done and revealed a normal intracardiac foetal heart with the advice to do post-natal echocardiography because the aortic arch was not seen clearly.
After delivery the baby was admitted to the neonatal intensive care unit (neonatal ICU) and connected to non-invasive ventilation. Echocardiography was done immediately after birth and revealed small atrial septal defect secundum type, two small muscular ventricular septal defects, bicuspid aortic valve with fusion between the right and non-coronary cusps, with mild aortic stenosis (peak instantaneous gradient of 45 mmHg and mean gradient of 25 mmHg), and a defect between the main pulmonary artery and ascending aorta. The aortic arch is left sided with no coarctation, and there is mild proximal right pulmonary artery stenosis.
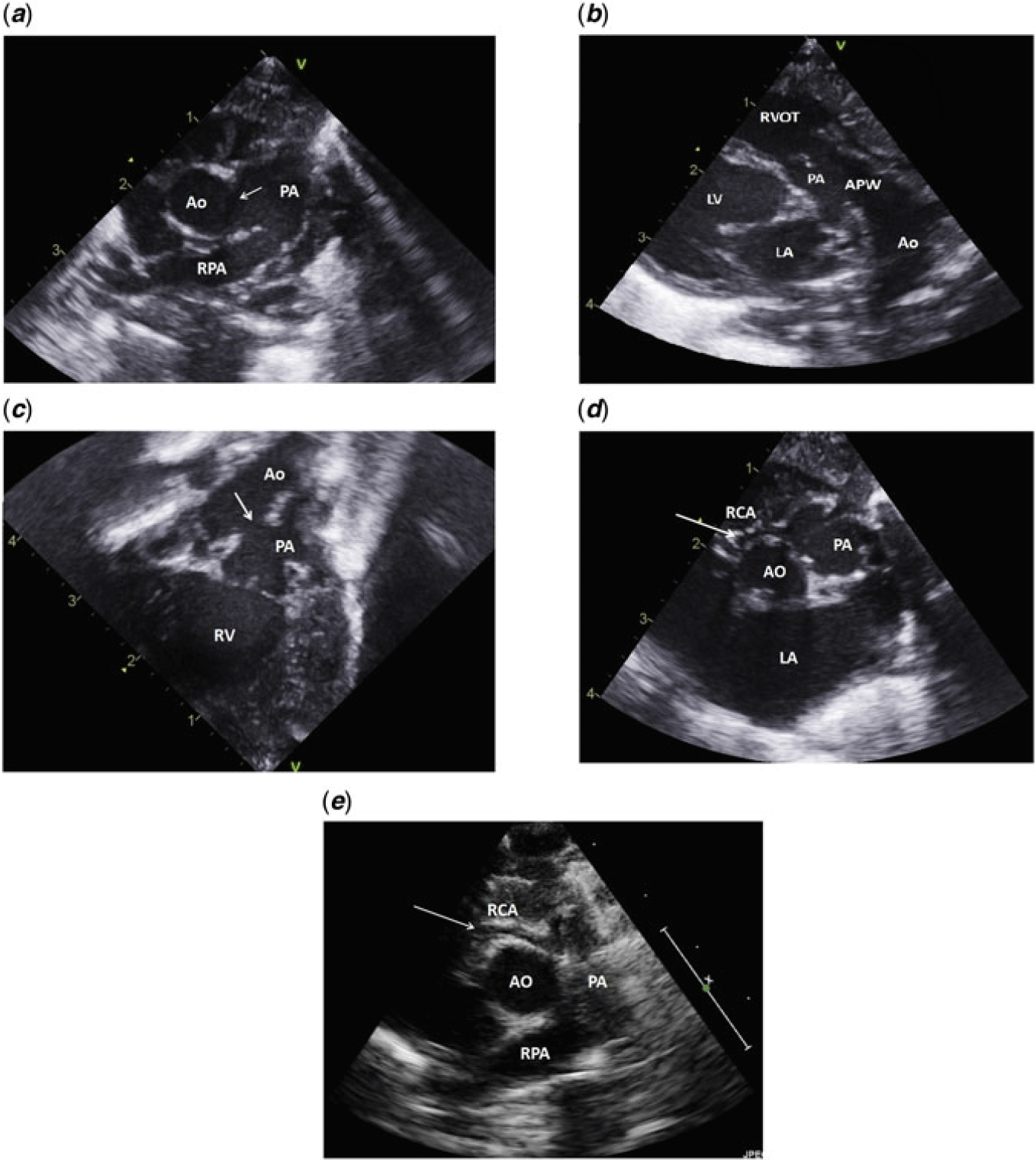
There was also mild tricuspid valve regurgitation, and mild dilatation of the right and left ventricles as well as the left atrium. Biventricular systolic function was good, and there was no pericardial effusion. The shunts at the atrial, ventricular and great vessel levels were left to right (Fig 1a–d).

Figure 1. Different echocardiographic views: (a) Short axis parasternal view showing the aortopulmonary window (white arrow). (b) Modified long axis view, showing the right ventricular outflow tract and main pulmonary artery. The aortopulmonary window is seen as a communication between the pulmonary artery and the aorta, mimicking a large patent ductus arteriosus. (c) Subcostal view with tilting of the probe anteriorly to visualise the right ventricular outflow tract showing the aortopulmonary window. (d) Short axis parasternal view showing the right coronary artery from the pulmonary artery. (e) Post-operative short axis parasternal view showing the patent right coronary artery. AO = aorta; APW = aortopulmonary window; LA = left atrium; LV = left ventricle; PA = main pulmonary artery; RCA = right coronary artery; RPA = right pulmonary artery; RVOT = right ventricular outflow tract.
Repeated follow-up echo revealed in addition to the previous findings that the right coronary artery is arising from the right sinus of the pulmonary artery and coursing towards the right ventricle.
The patient remained in the neonatal ICU for 2 months with an uneventful course. Feeding was built up gradually. He was given small doses of furosemide and spironolactone.
He was discharged home with a weight of 2 kg, to be readmitted at the age 4 months and 6 days for cardiac surgery. His weight at the time of surgery was 3 kg.
Surgery was done through median sternotomy and cardiopulmonary bypass. The surgical findings were large aortopulmonary window connecting the main trunks of the pulmonary artery with the ascending aorta. The right coronary artery was arising anomalously from the right pulmonary sinus.
The aortopulmonary window was closed using polytetrafluoroethylene (PTFE) patch. The right coronary artery was diverted to the aorta by suturing the patch to the sinus of the pulmonary artery and sandwiching the anterior aspect of the patch between the aorta and pulmonary arterial walls, avoiding sutures near branch pulmonary arteries. The baby was re-warmed and weaned from cardio-pulmonary bypass in the usual manner. The sternum was closed on the second post-operative day.
The ICU course was uneventful. He was extubated on the 5th post-operative day, and was discharged home on the 10th post-operative day.
Follow-up in paediatric cardiology clinic revealed that he was asymptomatic and developing appropriately with slow weight gain.
Last echo revealed no evidence of aortopulmonary window patch leak, bicuspid aortic valve with no stenosis or regurgitation, no left or right ventricular outflow tracts obstructions, no pulmonary insufficiency, good-sized right pulmonary artery, with proximal left pulmonary artery stenosis (peak instantaneous gradient of 25 mmHg), patent aortic arch and good biventricular systolic function with an ejection fraction of 65% (Fig 1e).
Discussion
Aortopulmonary window is a rare congenital cardiac defect characterised by abnormal communication between the ascending aorta and the pulmonary artery with two separate semilunar valves. Aortopulmonary window accounts for 0.1–0.2% of all congenital cardiac defects. It can occur in isolation or may be associated with complex cardiac malformations like interrupted aortic arch, ventricular septal defect, tetralogy of Fallot, or transposition of the great arteries.Reference Talwar, Agarwal and Choudhary1, Reference Najm, Jijeh, El Moazamy, Mufti, Abu-Sulaiman and Mutairi2
Aortopulmonary window may be classified into three types: type I (proximal) defects in which there is a defect between the proximal ascending aorta and the main pulmonary artery; type II (distal) defects, in which the defect is between the proximal right pulmonary artery and the aorta, and type III in which the defect is a combination of types I and II defects.Reference Mori, Ando, Takao, Ishikawa and Imai3
The current surgical outcome of the patients with aortopulmonary window is excellent, even when surgery is performed at the age of more than 3 months.Reference Kumar, Singh, Thingnam, Mishra and Jaswal4
Anomalous origin of the right coronary artery from the pulmonary artery (ARCAPA) associated with the aortopulmonary window is a very rare entity, with few reported cases in the literature.Reference Bhat, Chandrashekar, Mallikarjun and Girish Gowda5
ARCAPA is extremely rare with an incidence of 0.002% in the general population.Reference Yamanaka and Hobbs6 Sixty-six per cent of the cases of ARCAPA are associated with other congenital disorders such as aortopulmonary window, tetralogy of Fallot, ventricular septal defect, and aortic stenosis.Reference Williams, Gersony and Hellenbrand7
Since right ventricular oxygen demand is lower than that of the left ventricle, ventricular ischemia in ARCAPA is felt to be less common than in anomalous left coronary artery from the pulmonary artery (ALCAPA). This condition is well tolerated in the neonatal period due to the high pulmonary vascular resistance.Reference Mintz, Iskandrian, Bemis, Mundth and Owens8
The presence of aortopulmonary window in association with anomalous coronary artery will maintain the high pulmonary artery pressure so that the coronary artery flow through the anomalous coronary artery from the pulmonary artery will be maintained and myocardial ischemia will be less likely to develop.
Anomalous origin of both right and left coronary arteries from a single ostium from the anterior sinus of the pulmonary artery along with aortopulmonary window was also reported.Reference Singh, Loona, Suryavanshi, Sahoo and Mahant9
The surgical repair of ARCAPA associated with aortopulmonary window is mainly by intrapulmonary baffle using a patch to connect the right coronary artery origin to the aorta through the aortopulmonary window, similar to Takeuchi procedure, in addition to closing the aortopulmonary window.
A case report of aortopulmonary window associated with ARCAPA revealed a satisfactory patient condition with good cardiac function 24 years after surgical repair employing the Takeuchi procedure. The angiographic control obtained 5 years after the operation showed an optimal surgical result.Reference Gabbieri, Guadalupi and Stefanelli10
Anomalous origin of left coronary artery from the pulmonary artery in association with aortopulmonary has also been described. The left coronary artery arose from the right pulmonary artery in association with type III aortopulmonary window and interrupted aortic arch.Reference McMahon, DiBardino, Ündar and Fraser11
Conclusion
Aortopulmonary window is a rare congenital heart lesion, often associated with other CHDs. ARCAPA is the most commonly described coronary artery anomaly in association with aortopulmonary window. This report and the previous reports highlight the importance of very careful assessment of the coronary arteries in patients with aortopulmonary window.
Declaration of Patient Consent
The authors certify that they have obtained the appropriate patient’s family consent. They were informed that anonymous echocardiographic images will be posted in the journal.
Financial Support
This research received no specific grant from any funding agency, commercial or not-for-profit sectors.
Conflict of Interest
None.
Ethical standards
The study was approved by the institutional ethical and research committee.
Supplementary Material
To view supplementary material for this article, please visit https://doi.org/10.1017/S1047951119002543