


Introduction
Nonbiting midges of the genus Labrundinia Fittkau (Diptera: Chironomidae: Tanypodinae) are minute Diptera (1.0–2.5 mm). Currently, this genus contains 14 species, all of which are found in the New World with the exception of Labrundinia longipalpis (Goetghebuer, 1921) (Ashe and O'Connor Reference Ashe and O'Connor2009; Silva et al. Reference Silva, Fonseca-Gessner and Ekrem2011). The genus was erected by Fittkau (Reference Fittkau1962) based on the Palearctic species L. longipalpis (original combination Tanypus longipalpis), which has immatures that live in a variety of unpolluted water bodies from small streams and ponds to lakes and bays (Silva et al. Reference Silva, Fonseca-Gessner and Ekrem2011). Morphology-based species identifications of Labrundinia are considerably difficult or even impossible, particularly for adults. The few diagnostic characters used to separate species demonstrate considerable intraspecific variation, making their application to one species difficult. For example, male abdominal coloration, an important diagnostic species character, has an extensive variation within the genus.
The descriptions of Labrundinia species currently available are based mainly on males and do not include diagnostic characters for larvae and pupae at the species level. Association of chironomid immature stages can be achieved by individual rearing of larvae and collecting cast larval and pupal skins to establish the associations between each life stage (e.g. Ekrem et al. Reference Ekrem, Willassen and Stur2007). This procedure is time-consuming, however, and is not always successful for species with special environmental requirements. Thus, life stage associations based on genetic similarity of short DNA fragments (so-called DNA barcodes) may be more effective for species identification. Several studies have recognised the benefit of DNA sequences for associating immature stages with adult stages (e.g. Hebert et al. Reference Hebert, Penton, Burns, Janzen and Hallwachs2004a; Thomas et al. Reference Thomas, Raharivololoniaina, Glaw, Vences and Vieites2005; Ekrem et al. Reference Ekrem, Stur and Hebert2010a; Stur and Ekrem Reference Stur and Ekrem2011; Silva et al. Reference Silva, Wiedenbrug, Oliveira, Trivinho-Strixino and Pepinelli2012). DNA barcoding has previously been used to differentiate six Australian species within another genus of the subfamily Tanypodinae (Procladius Skuse, Carew et al. Reference Carew, Marshall and Hoffmann2011).
Tanypodinae is the third most diverse subfamily after Chironominae and Orthocladiinae, respectively (Ashe and O'Connor Reference Ashe and O'Connor2009). Thienemann and Zavřel (Reference Thienemann and Zavřel1916) established this subfamily based on the immature stages and its monophyly is strongly supported, with Podonominae as its sister-group (Cranston et al. Reference Cranston, Hardy and Morse2012). Larval Labrundinia have modified head capsules that are adapted for a predatory lifestyle, as is found in most members of Tanypodinae. Comparative morphological studies on larval head capsules (Gouin Reference Gouin1959; Bryce and Hobart Reference Bryce and Hobart1972) indicate that the features distinguishing the Tanypodinae larvae from those of the other subfamilies were regarded as adaptations for predation.
DNA barcoding employs standardised genomic fragments to enable species identification and discovery of cryptic taxa (Hebert et al. Reference Hebert, Cywinska, Ball and DeWaard2003; Kress et al. Reference Kress, Wurdack, Zimmer, Weigt and Janzen2005; Savolainen et al. Reference Savolainen, Cowan, Vogler, Roderick and Lane2005). Supporters of this technique argue that a short standardised fragment of DNA can be used to recognise taxa as well as increase the speed, objectivity, and efficiency of species identification (Meyer and Paulay Reference Meyer and Paulay2005). Initial tests of genetic barcoding using mitochondrial markers on animals have shown that a 658-base-pair fragment of the mitochondrial gene, cytochrome c oxidase subunit I (COI) is usually effective as a barcode sequence, providing more than 95% species-level resolution (Hebert et al. Reference Hebert, Cywinska, Ball and DeWaard2003, Reference Hebert, Penton, Burns, Janzen and Hallwachs2004a; Hajibabaei et al. Reference Hajibabaei, Janzen, Burns, Hallwachs and Hebert2006; Smith et al. Reference Smith, Woodley, Janzen, Hallwachs and Hebert2006). Nevertheless, despite these highly encouraging results, the success rate of this approach relies on the delineation between intraspecific and interspecific genetic divergence (Hebert et al. Reference Hebert, Stoeckle, Zemlak and Francis2004b; Meyer and Paulay Reference Meyer and Paulay2005). On the other hand, intraspecific and interspecific DNA sequence variation depends directly on the evolutionary and biogeographical relationships of the group in question, which might differ severely from one population or region to another (Kirkendale and Meyer Reference Kirkendale and Meyer2004).
Regarding the identification of insects, DNA barcoding has a number of drawbacks (Virgilio et al. Reference Virgilio, Backeljau, Nevado and Meyer2010). Recent speciation, incomplete lineage sorting, interspecific hybridisation, and infection by endosymbiotic bacteria such as Wolbachia Hertig (Rickettsiaceae) (Funk and Omland Reference Funk and Omland2003; Whitworth et al. Reference Whitworth, Dawson, Magalon and Baudry2007; Foottit and Adler Reference Foottit and Adler2009) may all negatively affect the performance of DNA barcoding in insects (Virgilio et al. Reference Virgilio, Backeljau, Nevado and Meyer2010). Undoubtedly, and perhaps more importantly, the reliability of DNA barcoding in insects might be challenging given their immense diversity (Foottit and Adler Reference Foottit and Adler2009), which affects the comprehensibility of DNA barcode sequence libraries to adequately represent the immense diversity in insects (Virgilio et al. Reference Virgilio, Backeljau, Nevado and Meyer2010).
In this study, we evaluate the feasibility of using DNA barcodes for species delimitation in Labrundinia using the standard barcode fragment of COI amplified with the universal primers of Folmer et al. (Reference Folmer, Black, Hoeh, Lutz and Vrijenhoek1994), LCO1490 and HCO2198. We also investigate whether partial COI gene sequences can be employed to associate life stages of species within this genus. We choose to focus on species of Labrundinia because this genus is currently being revised by the first author and material of numerous species is available from different localities in São Paulo State, Brazil. All unnamed species referenced in this study will be described in a future taxonomic manuscript, and published elsewhere.
Materials and methods
The taxa included in this study were selected to represent as many of the known morphotypes in Labrundinia as possible. Field work was conducted in São Paulo State, Brazil, without any design to test sampling regime or spatial distribution. Larvae and pupae were collected from aquatic systems < 200 km apart, using a hand-net. Some larvae and pupae were isolated in small vials covered with nylon screen and reared in the laboratory to obtain emerged adults. Several samples of different aquatic macrophyte species were collected and placed in a plastic tray in order to rear specimens to adulthood. Immature chironomids were preserved in absolute ethanol while imagines were kept in slightly dilute ethanol (∼80–85%) to avoid breakage. More than one life stage was sequenced from all the included species (Appendix). Different morphotypes were recognised based on variation in all observable morphological traits such as colouration, genital structures, shapes of pupal thoracic horn, and larval claws.
Ethanol-preserved specimens were dissected under a stereo microscope, and the wings, one pair of legs, and the antennae were mounted in Euparal on microscope slides. DNA was extracted from the remaining body parts in a buffered solution with the enzyme proteinase-K. DNA extraction, polymerase chain reaction (PCR), and bi-directional sequencing were performed at the Canadian Centre for DNA Barcoding (Guelph, Ontario, Canada), using standard protocols (http://ccdb.ca/pa/ge/research/protocols). Other specimens were analysed in the molecular lab at the Norwegian University of Science and Technology Museum in Trondheim (Norway), where DNA extraction followed the tissue protocol using a GeneMole robot (MoleGenetics, Lysaker, Norway). Each PCR was made in a total volume of 25 μL and contained 2 μL DNA template (concentration not measured), 1 × Qiagen PCR Buffer (Tris-Cl, KCl, (NH4)2SO4, 1.5 mM MgCl2), 2.0 μM additional MgCl2, 0.8 mM of dNTPs, 0.4 μM of each of the suggested standard barcode primers (Folmer et al. Reference Folmer, Black, Hoeh, Lutz and Vrijenhoek1994) LCO1490 (5′-GGTCAACAAATCATAAAGATATTGG-3′) and HCO2198 (5′-TAAACTTCAGGGTGACCAAAAAATCA-3′), 1 unit of HotStarTaq DNA Polymerase (Qiagen, Oslo, Norway), and 14.3 μL of ddH2O. Amplifications for the COI region were performed in a thermocycler with an initial denaturation step of 95 °C for 15 minutes, followed by five cycles of 94 °C for 30 seconds, 45 °C for 30 seconds, 72 °C for 1 minute, followed by 35 cycles of 94 °C for 30 seconds, 51 °C for 30 seconds, 72 °C for 1 minute, and one cycle at 72 °C for 5 minutes, then held at 4 °C.
The PCR products were purified using ExoSAP-IT (USB Products, Affymetrix, Santa Clara, United States of America) following protocols recommended by the manufacturer. Purified products were sequenced in both directions using BigDye 3.1 (Applied Biosystems, Life-technologies, Oslo, Norway) termination reactions and analysed on ABI 3730 genetic analysers. Sequences were assembled and edited using DNA BASER Sequence Assembler 3.2.4 (Heracle BioSoft S.R.L., Pitesti, Romania), checked for stop-codons and aligned as translated amino acids using default ClustalW options (Thompson et al. Reference Thompson, Plewniak and Poch1999) as implemented in MEGA 5.03 (Tamura et al. Reference Tamura, Peterson, Peterson, Stecher, Nei and Kumar2011). The alignment was trivial as no indels were observed in the sequences.
Neighbour-joining (NJ) and maximum likelihood trees were produced in MEGA 5.03 (Tamura et al. Reference Tamura, Peterson, Peterson, Stecher, Nei and Kumar2011), using Kimura two-parameter (K2P) (Kimura Reference Kimura1980) and GTR + G + I (Lanave et al. Reference Lanave, Preparata, Saccone and Serio1984) models, respectively. Support for specific tree topologies was estimated by bootstrap analysis with 1000 pseudoreplicate data sets (Felsenstein Reference Felsenstein1985). Pairwise sequence divergences within and between genetic clusters were calculated under a K2P model in MEGA. The analysis of intraspecific and interspecific genetic distances were based on the K2P (Kimura Reference Kimura1980) and maximum composite likelihood Tamura et al. (Reference Tamura, Nei and Kumar2004) models and were as the best-fit models of nucleotide substitution and base frequencies also calculated with MEGA 5.03. Intraspecific and interspecific distances were plotted as a histogram using PAST version 2.14b (Hammer et al. Reference Hammer, Harper and Ryan2001). A summary of species sequenced and their respective voucher and GenBank accession numbers is provided in the Appendix. All specimens and DNA barcodes are deposited in the Barcode of Life Data Systems (boldsystems.org, Ratnasingham and Hebert Reference Ratnasingham and Hebert2007) in the project Neotropical Tanypodinae (NEOTA).
Results
Partial COI gene-sequences were obtained from 79 specimens of 13 species (Appendix). The Folmer-primers worked well on templates from all tested species and no difference was observed in amplification or sequencing success with regard to different life stages. The aligned sequences in the majority (98.7%) were 657 base pairs long with 209 variable sites (31.8%), of which 200 (95.6%) were informative. Most variable sites occurred in the third codon-position (Table 1). The sequences were heavily AT-biased, specifically in the third position, which exhibited a combined average AT-composition of 89.9% (Table 1). A hierarchical likelihood ratio test of aligned sequences in MEGA 5.03 returned the general time reversible model with a parameter for invariable sites and gamma correction for rate heterogeneity (GTR + G + I) as the best model (−ln L = 4110.404, BIC =9991.709, AIC = 8531.870).
Table 1 Variable and informative sites and average nucleotide composition in the analysed COI gene sequences
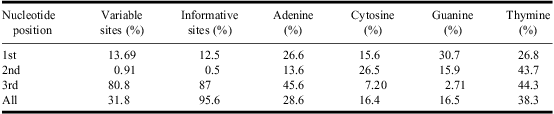
COI, cytochrome c oxidase subunit I.
Based on clustering of exemplars in the NJ analysis we were able to identify 12 new, undescribed Labrundinia species. In addition, Labrundinia tenata, previously described by means of morphology (Roback Reference Roback1987; Silva and Fonseca-Gessner Reference Silva and Fonseca-Gessner2009) was successfully sequenced and DNA barcodes were produced from all life stages.
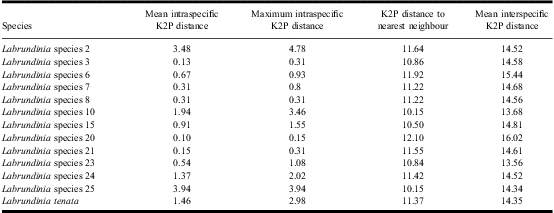
The pairwise distances for the analysed Labrundinia specimens, produced by both K2P and maximum composite likelihood models, showed distinctly larger interspecific than intraspecific divergences. Thus, there were distinct barcode gaps (Fig. 1). There were no identical gene sequences between species, and all species were distinguishable by genetic distances. Average intraspecific and interspecific K2P-distances for all analysed Labrundinia species were 0.91% and 14.53%, respectively. Maximum intraspecific divergence was observed in Labrundinia species 2 (4.78%), followed by Labrundinia species 25 (3.94%) and Labrundinia species 10 (3.46%) (Table 2). The lowest interspecific distances were found between Labrundinia species 10 and Labrundinia species 25 (average 10.8%), followed by Labrundinia species 10 and Labrundinia species 15 (average 11.23%). Intraspecific and interspecific distances produced by the maximum composite likelihood model provided similar results.

Fig. 1 Histogram of the calculated intraspecific and interspecific distances of partial COI sequences for the analysed Labrundinia specimens: (A) Kimura two-parameter. (B) Maximum composite likelihood.
Table 2 Summary of intraspecific and interspecific Kimura two-parameter distances between morphological species of the genus Labrundinia
The specimens identified morphologically as L. tenata were divided into two separate barcode clusters. Nucleotide sequences of these specimens differed by a minimum of 2.5% and in up to 22 nucleotide sites, but there were no observable morphological characters that differentiated the specimens belonging to theses clusters. Specimens of L. tenata from the two clusters were collected from four ecosystems in São Paulo State, three in São Carlos municipality, and one in Luiz Antonio municipality (Appendix). All specimens from one cluster were collected at the São Carlos Ecological Park, so there was some geographical structure in the clustering even though the localities in São Carlos are not more than 8.5 km from each other. Similarly, the specimens identified as Labrundinia species 10 also formed two distinct groups, but no morphological differences were observed. Nucleotide sequences of these specimens diverged by a minimum of 2.7% and up to 25 nucleotide sites. In this case, all specimens were collected in the same geographical locality (Ecological Park/Monjolinho stream).
DNA barcode sequences also indicated that three of the clusters were related species. This was subsequently confirmed by morphological analysis of the immature stages: Labrundinia species 8 resembled Labrundinia species 6 in the presence of a serrated claw in the larva and in the male abdominal colouration, but differed in the pupal thoracic horn ratio (length/width) that was higher in Labrundinia species 8. Nucleotide sequences of these specimens differed by minimum 15.6% and in up to 90 nucleotide sites. Labrundinia species 23 and Labrundinia species 25 were similar to Labrundinia species 2 in the male abdominal colouration, but diverged distinctly in the size and shape of the pupal thoracic horn, and in the number of spines that constitutes the lateroventral spine group (LVS) of the larval head. Nucleotide sequences of these specimens differed by minimum of 12.6% and in up to 133 nucleotide sites.
A NJ tree demonstrated that a substantial barcode divergence exists between all the analysed species of Labrundinia. For easier comparison with other DNA barcode studies, we here present the NJ trees produced by using the K2P model (Fig. 2). The NJ analysis, using maximum composite likelihood, yielded identical NJ trees, whereas trees resulting from maximum-likelihood analysis (using K2P and GTR + G + I models of substitution) only differ in the placement of Labrundinia species 25, which groups with Labrundinia species 10 as its closest cluster (data not shown). Boostrap support exhibited minor variation among all analyses, but were always 98% or higher for all species clusters.
Fig. 2 Neighbour-joining tree based on partial COI sequences (DNA barcodes) and the Kimura two-parameter substitution model. Numbers on branches are bootstrap values >70%. DNA barcodes enabled association of different life stages, which are labeled on the terminals. Colours denote different species.
Fig. 2 Continued
Discussion
DNA barcoding focuses on species delineation and identification, and not on phylogenetic inference. Moreover, COI has been found to be too variable for reliable phylogenetic analysis in Chironomidae (Ekrem et al. Reference Ekrem, Willassen and Stur2007, Reference Ekrem, Willassen and Stur2010b). However, the NJ tree based on analysis of pair-wise COI distances (Fig. 2) can be useful as a graphical representation of the genetic differences between sequences and clusters of sequences in the dataset. In this tree (which is not a representation of the most probable phylogeny of the included taxa), it is noticeable that there is a substantial barcode divergence between all the analysed species of Labrundinia.
The use of DNA barcodes also enables the association of different life stages based on genetic similarity of phenotypic characteristics (Hebert et al. Reference Hebert, Cywinska, Ball and DeWaard2003). In many chironomids, descriptions and diagnoses are largely or entirely based on a single life stage or even a single sex of a life stage. Consequently, incomplete knowledge of the life stages of a species precludes the use of morphological characters and natural history information that might be of particular interest for testing ecological, phylogenetic, and evolutionary hypotheses. The successful life stage associations made in this, as well as previous studies (Carew et al. 2005, Reference Carew, Pettigrove, Cox and Hoffmann2007, 2011; Sinclair and Gresens Reference Sinclair and Gresens2008; Wiedenbrug et al. Reference Wiedenbrug, Mendes, Pepinelli and Trivinho-Strixino2009; Ekrem et al. Reference Ekrem, Stur and Hebert2010a; Stur and Ekrem Reference Stur and Ekrem2011; Silva et al. Reference Silva, Wiedenbrug, Oliveira, Trivinho-Strixino and Pepinelli2012), show that similar success may be expected in the association of adults and immatures of other chironomids (and other insects), which enables a complete taxonomic description of species where rearing is difficult or even impossible.
We observed low levels of intraspecific divergence within the species analysed. This result is perhaps not surprising given that most of the specimens from a certain species were sampled in the same locality. Nevertheless, our results clearly demonstrate the potential of using DNA barcodes in the identification of chironomids, at least in a local geographical scale. Moreover, the average intraspecific variation reported here is similar to other studies of insects. Average intraspecific variation of 0.9% (Ekrem et al. Reference Ekrem, Willassen and Stur2007) and 2.32% in Chironomidae (Sinclair and Gresens Reference Sinclair and Gresens2008); 2.76% (Hebert et al. Reference Hebert, Penton, Burns, Janzen and Hallwachs2004a) and 0.46% (Hajibabaei et al. Reference Hajibabaei, Janzen, Burns, Hallwachs and Hebert2006) in Lepidoptera; and 1.1% in mayflies (Ball et al. Reference Ball, Hebert, Burian and Webb2005) have been recorded. The aforementioned results indicate that a single threshold value for species delimitation is not appropriate. Even though recent efforts have been made to overcome this drawback (e.g. Automatic Barcode Gap Discovery) for primary species delimitation (Puillandre et al. Reference Puillandre, Lambert, Brouillet and Achaz2011), character-based methodologies (DeSalle et al. Reference DeSalle, Egan and Siddall2005; DeSalle Reference DeSalle2007), and Barcode Index Numbers (Ratnasingham & Hebert, Reference Ratnasingham and Hebert2013), additional studies will be required to determine the best method for species delineation using DNA barcodes.
According to Aliabadian et al. (Reference Aliabadian, Kaboli, Nijman and Vences2009), the barcode gap, i.e., the difference between intraspecific and interspecific distances, allows for identification success in distance-based barcoding. Our data show that the distribution of intraspecific and interspecific divergences in Labrundinia exhibit a clear barcode gap which may be considered as a predictor of the identification success. Nonetheless, DNA barcodes were not enough for precise identification in all cases. For example, L. tenata specimens separated into two distinct groups, having nucleotide sequence divergences up to 2.5% and no observable morphological differences. Based on the suggested threshold of 2–3% applied to distinguish species (Hebert et al. Reference Hebert, Cywinska, Ball and DeWaard2003), the two clusters can be considered one species. Labrundinia species 10 exhibited similar results. Using DNA barcodes, Ekrem et al. (Reference Ekrem, Willassen and Stur2007) also obtained two separate clusters of Micropsectra notescens (Walker) (Diptera: Chironomidae: Chironominae) specimens, even though they were unable to find any distinct morphological differences separating the clusters. In contrast, Sinclair and Gresens (Reference Sinclair and Gresens2008), found one cluster represented by two species of Cricotopus van der Wulp (Diptera: Chironomidae: Orthocladiinae). The specimens had <3% COI sequence divergence, but presented consistent morphological differences. These results indicate that we need further studies using nuclear markers and morphological and ecological data in order to elucidate relationships between specimens that currently appear to belong to L. tenata and Labrundinia species 10.
DNA barcodes are argued to be valuable in species identification of taxonomic groups that are difficult to identify using morphology and Hebert et al. (Reference Hebert, Cywinska, Ball and DeWaard2003, Reference Hebert, Penton, Burns, Janzen and Hallwachs2004a) stressed that DNA barcoding permits the assignment of unidentified specimens to known species as well as to identify species new to science. Our findings ratify this assertion as Labrundinia species 8, Labrundinia species 23, and Labrundinia species 25 were unveiled as new, undescribed species only after their significantly different and deeply divergent barcode clusters were discovered. These species differ mainly on the pupal thoracic horn, which seems to provide good diagnostic characters for many Labrundinia species, showing consistent patterns of interspecific variation. However, before the DNA barcode analyses, the pupal thoracic horn differences were treated as subtle morphological variation. Similarly, Carew et al. (Reference Carew, Marshall and Hoffmann2011) separated apparently cryptic species of Procladius Skuse (Diptera: Chironomidae: Tanypodinae) by means of morphological characters in the immature stages only after the analysis of the DNA barcode data. In a more general perspective, our results support DNA barcoding using COI as a promising approach for accurate interpretation of morphological variations within nonbiting midges in the subfamily Tanypodinae.
The DNA barcoding approach has been argued to be imprecise for consistent species delimitations by several authors (e.g. DeSalle et al. Reference DeSalle, Egan and Siddall2005; Will et al. Reference Will, Mishler and Wheeler2005; Rubinoff et al. Reference Rubinoff, Cameron and Will2006; Ebach Reference Ebach2011), mostly due to general methodological reservations. The presence of mitochondrial pseudogenes (Bensasson et al. Reference Bensasson, Zhang, Hartl and Hewitt2001) or incomplete lineage sorting may lead to species-level paraphyly and polyphyly (Funk and Omland Reference Funk and Omland2003), blurring delineation of species by monophyletic barcode clusters criterion (Ekrem et al. Reference Ekrem, Stur and Hebert2010a). Nevertheless, none of these drawbacks seems to be an issue using COI sequences in species-level identification of Chironomidae (Carew et al. 2005, 2007; Sinclair and Gresens Reference Sinclair and Gresens2008; Ekrem et al. Reference Ekrem, Willassen and Stur2010b; and this study).
Conclusions
In our study of Labrundinia species, analysis of DNA barcode sequences using NJ trees supported 13 cohesive species clusters, of which three similar groups were subsequently linked to distinct morphological characters in the immature stages. DNA barcodes also assisted in associating different life stages. A distinct barcode gap and considerable interspecific pairwise divergences were observed, which allowed for unambiguous identification of all analysed species. Moreover, although DNA barcodes worked well for species delimitation in Labrundinia, inclusion of sequence data from extra nuclear markers is recommended in order to strengthen these results. Finally, efforts should be made to obtain specimens of L. longipalpis from the Palearctic as knowledge of their genetic distance to the New World species and to North American populations of L. longipalpis may be essential to understand the species limits within the genus.
Acknowledgements
The authors thank Paul Hebert and the staff at the Biodiversity Institute of Ontario, University of Guelph, Canada for sequencing some of the specimens used in this study. Sequencing in Guelph was supported from Genome Canada through a grant by the Ontario Genomics Institute to Paul Hebert. The authors are indebted to Mateus Pepinelli for his assistance, processing some samples in Canada. Thanks also to Caroline Silva Neubern de Oliveira, Sofia Wiedenbrug, and Susana Trivinho Strixino for providing us with important material. Three anonymous reviewers offered valuable comments on this manuscript. F. L. Silva received financial support from the National Council for Scientific and Technological Development (CNPq proc. 141092/2009-2). Financial support was also given by the Research Council of Norway (RCN), through a Personal Mobility Grant.
Appendix List of analysed specimens with associated sample localities in São Paulo State, Brazil, voucher reference numbers and GenBank accessions. ‘‘FA’’ indicates samples processed by Fabio Laurindo da Silva; ‘‘UFSCAR FL’’ indicates samples processed by Mateus Pepinelli




