


Introduction
Understanding the dynamics of invertebrate predator-prey interactions in their natural environment can be challenging. Within arable systems, knowledge of the links between pest species and their predators is fundamental for understanding species interactions and food webs. Traditional methods for analysing predation, such as microscopic identification of prey remains or direct observation of predation, are of little use for identifying trophic links in natural environments, particularly for small invertebrates, which are often nocturnally active fluid feeders. The application of molecular markers to elucidating predation has been a fast growing field (Symondson, Reference Symondson2002; King et al., Reference King, Read, Traugott and Symondson2008) and allows detection of predation with high sensitivity and specificity.
However, like all molecular methods, the molecular detection of predation is prone to potential sources of error, giving rise to both false positive and false negative results. Four main sources of error have so far been investigated. Using PCR-based methods, it is not possible to distinguish active predation from scavenging on dead prey. Both Foltan et al. (Reference Foltan, Sheppard, Konvicka and Symondson2005) and Juen & Traugott (Reference Juen and Traugott2005) showed that DNA from decaying prey was readily detected in predator guts. Secondary predation is also a possible source of error. Sheppard et al. (Reference Sheppard, Bell, Sunderland, Fenlon, Skervin and Symondson2005) showed that it was possible to detect aphid DNA in carabid beetles that had fed on spiders that had been digesting aphid DNA for up to 4 h. Additionally, soil dwelling invertebrates living in close proximity to their food sources may become surface contaminated with DNA from that food source (Remén et al., Reference Remén, Krüger and Cassel-Lundhagen2010). These three sources of error can potentially lead to false positive results. DNA extracted from predator gut contents or faecal samples can often contain PCR inhibitors (Hebert et al., Reference Hebert, Darden, Pedersen and Dabelsteen2011), and this can lead to false negatives (Juen & Traugott, Reference Juen and Traugott2006). The now routine screening of predator DNA extracts with general or species specific PCR primers and using PCR facilitators such as bovine serum albumin (Juen & Traugott, Reference Juen and Traugott2005, Reference Juen and Traugott2007) has largely eliminated this source of false negatives.
King et al. (Reference King, Read, Traugott and Symondson2008) also suggested that field methods that either confine the sample in a small area (e.g. dry pitfall traps, sweepnets, malaise traps) or cause the animal to regurgitate its stomach contents through submersion in a killing solution (e.g. wet pitfall traps, hand collecting into ethanol), could also lead to false positive results. Small predators are effectively collected using suction sampling (Sunderland et al., Reference Sunderland, Powell, Symondson and Jervis2005), providing measures of density and potentially a source of predators for gut content analysis. These samplers take the form of either a portable G-vac (modified garden blowers, less commonly Dietrick (D-vac) samplers) that use a net or sock to collect the sample, or a Vortis machine that uses centrifugal force (Wheater et al., Reference Wheater, Bell and Cook2011). As the predators and target prey combine in the collecting cup or bag, two potential sources of false positives may occur. It is possible that because of the impact of the high-speed collection methods, small, fragile invertebrates such as aphids and collembola become squashed and then ectopically contaminate predators with their DNA. Additionally, due to containment in the enclosed environment of the collecting cup or bag, predation could be ‘forced’, allowing individual predators to forage freely on captured prey, especially immediately after the machine has been turned off. Such predation could potentially inflate the number of predators testing positive and lead to trophic links that do not occur naturally in the field. It was suggested that low vortis/vacuum pressures and transferring predator immediately onto ice, could mitigate these problems (King et al., Reference King, Read, Traugott and Symondson2008).
Recent studies have investigated the potential for predator collection methods to give rise to false positive results. Harwood (Reference Harwood2008) and Chapman et al. (Reference Chapman, Romero and Harwood2010) compared hand collection of spider predators with sweep-netting and Vortis sampling, respectively. In both studies, no significant differences were found between the proportions of hand collected versus sweep-netting/vacuum collected spiders testing positive for Diptera protein (Harwood, Reference Harwood2008) or aphid and collembolan DNA (Chapman et al., Reference Chapman, Romero and Harwood2010), suggesting that these alternative sampling methods were suitable for the collection of predators for molecular analyses. Conversely, Greenstone et al. (Reference Greenstone, Weber, Coudron and Payton2010) found moderate levels of false positives in predators (pentamid bugs and chrysomelid beetles) collected using both rough (predators beaten from plants onto cloth and collected using a pooter) and hand collection methods. They suggested that the difference between their results, and those of Harwood (Reference Harwood2008) and Chapman et al. (Reference Chapman, Romero and Harwood2010), could be due to differences in predator biology.
Here, we test the hypothesis that suction sampling of predators for molecular analyses leads to high levels of false positive trophic links. We designed a set of complementary experiments using field-based techniques and starved spider and beetle predators (hence, two very different feeding modes and biology). The predators were screened by PCR for the presence of multiple prey, revealing trophic errors that are most likely to occur in studies that use suction samplers.
Materials and methods
Predators were collected from the field and then starved for seven days prior to the start of the experiments, to ensure their guts were empty. They were marked with acrylic paint to identify starved from any non-starved predators that might be simultaneously collected from the field during the experiments. Both experiments were conducted in winter wheat fields.
Field methods – experiment 1
Using a Vortis sampler (Burkhard Ltd, Rickmansworth, UK), 20 aphids (Sitobion avenae) per sample (n=12 samples) were sucked from a plastic disc into the Vortis along with one Tachyporus hypnorum (Staphylinidae), one Bembidion lampros and one Demetrias atricapillus (both Carabidae). A further nine sucks of three seconds each was performed across a winter wheat field, replicating a standard protocol (overall, ten sucks each lasting three seconds, equating to a sampling area of 0.6 m2: Bell et al., Reference Bell, King, Bohan and Symondson2010). At this point, in each replicate sample, the D. atricapillus was removed from the collection pot, placed in an individual 1.5 ml microcentrifuge tube and frozen at –80°C (a priori removal). A starved Notiophilus biguttatus (Carabidae) was then added (post-hoc addition) to the sample before all of the contents were transferred into a plastic bag. Bags were placed in an insulated box with ice packs (temperature approximately 5°C) in the field and then transferred to an incubator with no light and set at 10°C. Predators were killed in absolute ethanol after 1 h and 24 h (six replicates each). The removal of D. atricapillus was used to assess whether DNA from squashed aphids or collembola could coat the beetles’ exoskeleton. The post-hoc addition of N. bigutattus was used to assess whether false positives could be caused by predation in the collection cup, independent of the effect of the Vortis. Positive results for T. hypnorum or B. lampros could be due to either ectopic contamination or post-collection predation.
Field methods – experiment 2
Using a converted Electrolux McCulloch BVM 250 leafsucker with a muslin collection sock, starved spiders (four each of Bathyphantes gracilis, Tenuiphantes tenuis (both Linyphiinae), Pachygnatha degeeri (Tetragnathiidae) and Erigone spp. (Erigoninae)) were released and immediately sucked directly from the soil surface (15 s suck at full power over an area of 0.5 m2: Davey, Reference Davey2010). The leafsucker was switched to low power to position it over a 10×10×30 cm plastic box 25% full of dry ice. The surface temperature of dry ice is approx −75°C and, therefore, sufficiently cold to kill specimens almost immediately, precluding feeding at this stage. The leafsucker was turned off, and the contents of the collection sock were immediately emptied into the box containing dry ice. The experiment was performed twice, once at the field margin and once 10 m into the field. In both experiments, in addition to the starved predators, many other invertebrates (including Diptera, Collembola, Coleoptera, Hemiptera, Arachnida), soil, grit and wheat straw were sucked from the soil.
Molecular methods
Genomic DNA was extracted using DNeasy Tissue Kits (Qiagen, Crawley, UK) following the manufacturers instructions. Predators (except D. atricapillus from experiment 1) were partially crushed in 200 μl of extraction buffer (180 μl Buffer ATL and 20 μl Proteinase K, both Qiagen) prior to the 2-h incubation stage. DNA was extracted from D. atricapillus without prior crushing (the aim was to test for topical contamination). To check for potential cross contamination of samples during DNA extraction, each batch of extractions included a negative control (a ‘blank sample’ extracted using the same protocol as invertebrate samples: King et al., Reference King, Read, Traugott and Symondson2008).
All samples were initially screened with the universal primers LCO1490 and HCO2198 (Folmer et al., Reference Folmer, Black, Hoeh, Lutz and Vrijenhoek1994) that amplify a 710 bp fragment of the mitochondrial cytochrome oxidase subunit I (COI) gene. This was to check for successful extraction of amplifiable DNA. Predators were screened for prey DNA using the primers listed in table 1. PCR reactions were carried out in a total volume of 25 μl, consisting of 1× PCR master mix (Invitrogen, Paisley, UK), 4 mM MgCl2 (Invitrogen), 0.05 mM dNTP mix (Invitrogen), 2.5 μg BSA (bovine serum albumin, New England Biolabs, Hitchen, UK), 0.1 μM each forward and reverse primer, 0.625 U Taq polymerase (Invitrogen) and 2.5 μl DNA. After an initial denaturing step at 94°C for 2 min 30 s, amplification proceeded for 35 cycles at 94°C for 30 s, annealing temperature (table 1) for 30 s, 72°C for 45 s and a final extension at 72°C for 10 min. PCRs were carried out in a MJ Research PTC200 thermal cycler (Bio-Rad, Hemel Hempstead, UK). PCRs were visualised on 1.5% (w/v) agarose gels stained with ethidium bromide.
Table 1. PCR primers used in this study.

Controlled feeding trial
To ensure that the starvation period was sufficient to empty the guts of the predators used in the above experiments, the detection limit for the Collembola spp. primers (Kuusk & Agustí, Reference Kuusk and Agustí2008) was determined using a controlled feeding trial with N. biguttatus as the predators. Adult N. biguttatus were collected from the soil surface in arable fields using an electric pooter and maintained under controlled conditions (16°C and a 16:8 L:D cycle) with access to water but no food. After seven days of starvation, beetles were placed individually in 5 cm Ø Petri dishes with three adult Folsomia candida. After 2 h, beetles that had consumed all three F. candida were removed and placed individually into clean Petri dishes. Beetles were killed at −80°C at set times post-feeding period (1, 3, 6, 12 and 24 h after the mid-point of the feeding period). DNA was extracted from crushed whole beetles using the PureGene Tissue Extraction Kit (Gentra, Minneapolis, MN, USA). All beetles were screened for the presence of collembolan DNA using primers Col4F and Col5R (Kuusk & Agustí, Reference Kuusk and Agustí2008).
Data analysis
For experiment 1, the proportions of each predator (excluding D. atricapillus) testing positive for aphid or collembolan DNA were modelled as a binomial generalized linear model whilst accounting for differences between prey species and time periods using Genstat 13th Edition (VSN international Ltd). For the Notiophilus/Isotoma feeding trial, the median detection time (equivalent to the detectability half-life of Chen et al. (Reference Chen, Giles, Payton and Greenstone2000)), i.e. the time at which 50% of beetles still tested positive for collembolan DNA, was determined using Probit analysis performed in MINITAB v 15 (Minitab Inc., 2008).
Results
Field experiment 1
Of the 48 predators used in this experiment, five (two B. lampros and three T. hypnorum; table 2) were not recovered from the collection pot in the Vortis suction sampler. Of the 12 D. atricapillus removed immediately after Vortis sampling, four (33%) tested positive for aphid DNA and two (17%) for collembolan DNA, indicating high rates of topical contamination.
Table 2. Results of Experiment 1 using beetle predators. A ‘+’ and ‘–’ indicate that a predator tested either positive or negative, respectively for a particular prey. An ‘x’ indicates that a predator was not present in the Vortis collection cup.

a – Demetrias atricapillus was removed from all replicates and killed prior to storage in an incubator. For clarity, results for this species are still presented with the replicate from which they were removed.
Of the 12 N. biguttatus added after completion of Vortis sampling, three (25%) tested positive for aphids and six (50%) for collembola, demonstrating high rates of predation within the sample bags during storage. The median detection time for the N. biguttatus fed with F. candida in the feeding trial was 6.39 h (fig. 1). No beetles tested positive in either the 12 h or 24 h post-feeding time periods, showing that the starvation period was sufficient to ensure that beetles could not have had detectable collembolan DNA within their guts at the time the experiments were carried out.

Fig. 1. Detectability of collembolan DNA in the gut of Notiophilus biguttatus. ▪, PCR data. Lines are fitted probit models with 95% confidence intervals. The median detection time (the time at which 50% of beetles test positive) is 6.39 h.
A combination of contamination and/or predation was shown through analysis of the other two predators, T. hypnorum and B. lampros, both of which showed positives for aphid and collembolan DNA (table 2). Two B. lampros and three N. biguttatus tested positive for both aphid and collembola at the same time. However, no significant difference was found between the proportion of predators testing positive for collembolan and aphid DNA (t=0.68, P=0.52), between the proportion of predators testing positive at 1 h and 24 h (t=–0.51, P=0.63) or any differences in overall prey DNA detection between B. lampros compared and either N. biguttatus (t=–0.40, P=0.70) or T. hypnorum (t=–0.48, P=0.64).
Field experiment 2
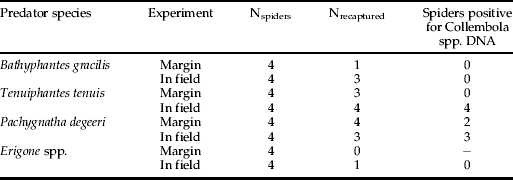
Nineteen of the 32 starved spiders used in this experiment were recovered. No individuals of B. gracilis and Erigone spp. tested positive for the presence of collembolan DNA (table 3). However, despite using a methodology that did everything possible to prevent post-vacuum sampling predation, four T. tenuis and five P. degeeri tested positive for collembolan DNA (table 3).
Table 3. Results of Experiment 2 using spider predators, showing recapture rates and numbers of predators testing positive for collembolan DNA
Discussion
Contrary to the conclusions of Chapman et al. (Reference Chapman, Romero and Harwood2010), the results from these two experiments show that suction sampling has the potential to give rise to high numbers of false positive results. This could lead to inflation of the number of predators scored as positive for a particular prey species and/or a misrepresentation of the prey spectrum of specific predators. It is recommended that appropriate experiments, similar to those conducted here and by Chapman et al. (Reference Chapman, Romero and Harwood2010) and Greenstone et al. (Reference Greenstone, Weber, Coudron and Payton2011), be conducted in order to determine the field sampling methods that best minimise the rates of false positive results in all analyses of the molecular detection of predation.
In experiment 1, six D. atricapillus tested positive for aphid and collembolan DNA. As individuals of this species were placed immediately in ethanol after Vortis sampling and were not crushed during DNA extraction, the positive results were almost certainly due to ecotopic contamination of aphid and collembolan DNA on the beetles’ exoskeleton. Greenstone et al. (Reference Greenstone, Weber, Coudron and Payton2011) found that a high proportion of Podisus maculiventris and Coleomegilla maculata, deliberately surface contaminated with DNA of Leptinotarsa juncta, tested positive for this species. However, if these beetles did manage to eat target prey within this very short window of opportunity (a few seconds) then we cannot entirely rule out detection of gut contents, as demonstrated by Pons (Reference Pons2006) who were able to extract prey DNA from entire tiger beetles. Individuals of N. biguttatus also tested positive for predation on both aphids and collembola. As this predator species was added after the end of the vortis period, these positive results can only be due to active predation post vortising. The positive results for B. lampros and T. hypnorum could be due to either ectopic contamination or in-cup predation. In experiment 2, despite being killed immediately after collection, nine spiders tested positive for collembolan DNA (table 3). As with B. lampros and T. hypnorum from experiment 1, this shows that either the spiders were contaminated ectopically with DNA from squashed collembola or that they were able to predate collembola within the collection sock of the modified leafsucker. Given the methodology employed in experiment 2, the latter seems unlikely.
The results for the 24-h time period for experiment 1 would suggest that, even at the low temperatures within the incubator, the beetle species are still able to capture and consume prey. Kruse et al. (Reference Kruse, Toft and Sunderland2008) have shown that temperature has a marked effect both on activity patterns and prey capture ability of Calathus fuscipes and Poecilus versicolor. These beetles had very low activity patterns at low temperature, but were able to capture Drosophila at temperatures below 10°C. Alternatively, it is possible that the beetles could still be testing positive after having eaten prey before being put at low temperature. It has been shown that the temperature at which controlled feeding trials are carried out has an effect on the median detection time of prey DNA within the predator's gut (Hoogendoorn & Heimpel, Reference Hoogendoorn and Heimpel2001; Davey, Reference Davey2010). Mean detection rates of S. avenae DNA in the gut of Pterostichus melanarius were significantly higher in feeding trials conducted at 12°C and 16°C compared to one performed at 20°C (von Berg et al., Reference von Berg, Traugott, Symondson and Scheu2008). For Folsomia candida fed the nematode Phasmarhabditis hermaphrodita, Read (Reference Read2007) found that lowering the temperature at which the feeding trial was conducted resulted in both an increase in median detection time (22.18 h at 24°C and 36.83 h at 4°C) and a lengthening of the maximum detection time (36 h at 24°C and 48 h at 4°C).
The results of experiment 2 show that, despite the short time between sample collection and placing the invertebrates on dry ice, some of the spiders were able to predate collembola within the collection sock. If the positive results were due to ectopic contamination with haemolymph from collembola damaged during suction sampling, we might expect positive results from all four species. Although sample numbers per species were too low to test for differences between spider species, the results potentially serve to highlight the opportunistic nature of predation in the case of both web-dependent (T. tenuis) and itinerant (P. degeeri) predators, even where precautions have been taken to minimise its occurrence.
In conclusion, the results of these experiments clearly show that mass sampling, using either Vortis samplers or converted leaf blowers, can lead to both contamination and/or unwanted predation within the equipment. Because food webs seem to be often characterised by many weak and few strong trophic links (Wooton & Emmerson, Reference Wootton and Emmerson2005), these sources of false predation have the potential to undermine predictive models of food web function. Experiments must be done prior to mass field collections of predators to find the method that best minimises the risk of false positive results. Some prey, such as aphids and collembola, may be relatively fragile and more likely to break up and contaminate predators externally. Where this is likely, gentler methods, such as hand sampling or using low pressure suction samplers, may be preferable for obtaining samples for DNA analyses. These techniques may underestimate the density of some species (Bell et al., Reference Bell, Wheater, Henderson, Cullen, Toft and Scharff2002); and, therefore, vacuum sampling can be used, in parallel, to estimate population densities. Other prey, such as the Aphrodes leafhoppers in the study by Virant-Doberlet et al. (Reference Virant-Doberlet, King, Polajnar and Symondson2011), are hard-bodied and survive vacuum sampling intact. In the latter study, some common spider species caught using a modified leaf blower never tested positive for Aphrodes DNA, while other spider species caught at the same time showed high rates of predation; if contamination were an issue, all species would have been contaminated. Such ‘internal’ controls can, thus, provide a useful way of monitoring for such problems and should be checked at an early stage before major fieldwork is undertaken.
Acknowledgements
We are grateful to Gwenaëlle Fournier and Fleur Moirot-Crouvisier for help in the field and to Rothamsted Research farm staff and Slade Farm, Glamorgan for accommodating our experiments. This work was part of a project entitled ‘Dynamic responses of predators to biodiversity in sustainable agriculture: spatial and molecular analyses’ funded by the Biotechnology and Biological Sciences Research Council (BBSRC grant BBD0011881). We thank two anonymous reviewers and the Subject Editor for providing valuable comments on an earlier draft of this manuscript.




