


Introduction
The soybean stalk weevil (SSW), Sternechus subsignatus Boheman, 1836 (Curculionidae: Sternechini), is a Neotropical pest in soybean agricultural systems that severely reduces yields in Brazil (Hoffmann-Campo et al., Reference Hoffmann-Campo, Oliveira, Mazzarin and de Oliveira1990). The first outbreaks were reported before 1968, in the State of Rio Grande do Sul, southern Brazil. High population densities were later found in Santa Catarina and Paraná, Brazil. SSW has also been reported in states of northeast Brazil, namely Bahia in the summer of 1997–1998 and in Maranhão in the summer of 2003–2004 (Hoffmann-Campo et al., Reference Hoffmann-Campo, da Silva and Oliveira1999; M. Meyer, personal observation). Outbreaks seem to be related to the expansion of soybean acreage in this region and to a lack of spatial and temporal non-diversity (Rosado-Neto, Reference Rosado-Neto1987).
In Argentina, another species from the same genus, S. pinguis (Fabricius, 1787) (Curculionidae: Sternechini), was detected in Tartagal Co., Salta Province, during the summer of 1987–1988 (Costilla & Venditti, Reference Costilla and Venditti1990) and in the Tucumán Province in 1998 (Salas et al., Reference Salas, Medina, Casmuz and Antoni2002). Both of these Sternechus species are distributed neotropically (Rosado-Neto, Reference Rosado-Neto1996). S. pinguis occurs in Venezuela, Surinam, French Guyana, Colombia, Ecuador, Peru, Bolivia and north Brazil (Amazonas and Pará states). S. subsignatus occurs in Paraguay, the central-west, south and southeast regions of Brazil, the northern Brazil state of Bahia and close to the Andes mountains in Bolivia and Argentina. However, it has yet to be determined whether SSW from both countries comprises one or two species. Molecular studies on genetic differentiation are useful for defining the degree of relatedness among genotypes, characterizing genotypes and delineating genetic distances among geographic populations. Such studies also reveal the abundance and distribution of genotypes within, and among, populations and help to define whether the distribution of genotypes is discrete or overlaps regions. Due to its small size, lack of recombination and presence of polymorphism, mtDNA is highly useful for population studies (Spanos et al., Reference Spanos, Koutroumbas, Kotsyfakis and Louis2000). It is also useful for clarifying differences between closely related species, or cryptic species, such as those within the lepidopteran family Hesperiidae (Hebert et al., Reference Hebert, Penton, Burns, Janzen and Hallwachs2004).
Although SSW is potentially devastating soybean pest, molecular genetic studies on this species are lacking. The purpose of this study was to determine intraspecific molecular polymorphism of geographically distinct Sternechus populations by studying mitochondrial and genomic DNA with Randomly Amplified Polymorphic DNA (RAPD) molecular markers. Our results shed light on pest invasion patterns and show that the SSW populations we studied constitute single species.
Materials and methods
Insect collection
SSW from different geographic locations (fig. 1) were sampled between December 2003 and February 2004 (table 1). Adults were hand collected from soybean fields. Specimens were kept stored at –20°C in dehydrated silica gel with moisture indicator. The silica gel was changed each time the indicator turned pink. Information on S. subsignatus collection localities is listed in table 1. DNA extraction was performed with a protocol based on CTAB salts (Rogers & Bendich, Reference Rogers, Bendich, Gelvin and Schilperoort1988). Voucher specimens are deposited in the entomology collection, ‘Pe. J.S. Moure’, Zoology Department, Federal University of Paraná, Curitiba, Brazil (DZUP).

Fig. 1. Soybean stalk weevil collection sites. Site names are described in table 1.
Table 1. Soybean stalk weevil collection data.
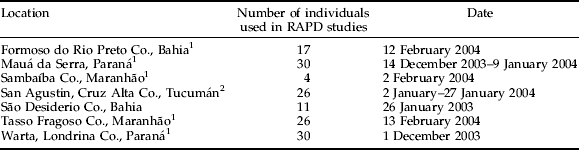
1 Brazilian state.
2 Argentinean province.
RAPD analysis
The DNA of 150 insects was individually extracted and diluted in TE buffer. RAPD reactions were performed in a PTC 200 thermal cycler (MJ Research, Inc., Watertown, MA, USA) using the following ten-mer Operon primers (Operon Technologies, Alameda, CA, USA) primers: A01 (5′-CAGGCCCTTC-3′), A09 (5′-GGGTAACGCC-3′), A16 (5′-AGCCAGCGAA-3′), A17 (5′-GACCGCTTGT-3′), B01 (5′-GTTTCGCTCC-3′), B05 (5′-TGCGCCCTTC-3′), C09 (5′-CTCACCGTCC-3′), C15 (5′-GACGGATCAG-3′), F03 (5′-CCTGATCACC-3′) and G13 (5′-CTCTCCGCCA-3′). We screened 25 different primers using pooled samples of DNA from each population and selected ten primers that amplified all the DNA samples and produced scorable bands that could be used to identify polymorphisms between the different individuals. DNA was amplified with 45 cycles of a thermal programme of 94°C for 15 s, 39°C for 30 s and 72°C for 1 min with a final extension of 72°C for 7 min. PCR amplification products were electrophoresed in 1.3% agarose gel with ethidium bromide, at 120 V, for 2.5 h in TBE buffer. Digitized photographs of gels generated from the RAPD analyses were obtained with a Kodak Electrophoresis and Documentation Analysis System 290 (Eastman Kodak Company, NY, USA). The presence or absence of RAPD products was coded in a binary matrix. A genetic similarity matrix was constructed based on RAPD data using the SIMQUAL module with the Dice coefficient. Clustering was performed with the unweighted pair-group method with arithmetic mean (UPGMA) from NTSYS-pc software (Rohlf, Reference Rohlf1993). The Dice coefficient was calculated as 2h/(a+b), where h is the number of matching bands and a+b is the total number of matching and non-matching bands being compared (Dice, Reference Dice1945). This coefficient assumes that bands of identical size are genetically homologous. Bootstrap analysis was performed with the Winboot programme using 1000 re-samplings (Yap & Nelson, Reference Yap and Nelson1996). Population structure studies were performed with analysis of variance of molecular data (AMOVA) using Arlequin Software (Schneider et al., Reference Schneider, Kueffer, Roessli and Excoffier2000). In the AMOVA analysis, sources of variation were divided into two levels: among geographic location, and among individuals within geographic locations.
PCR and sequencing analysis
Fragments of the cytochrome B (CytB) gene were amplified using the following primers CytBF (5′-GGACAAATATCATGAGGAGCAACAG-3′) and CytBR (5′-ATTACTCCTCCTAGCTTATTAGGAATTG-3′) (Monteiro et al., Reference Monteiro, Perz, Panzera, Dujardin, Galvão, Rocha, Noireau, Schofield and Beard1999). The PCR reaction was performed using a PTC 200 thermal cycler programmed for 34 cycles of 94°C for 1 min, 48°C for 1 min and at 72°C for 1.5 min with a final extension at 72°C for 5 min. The 539 bp PCR amplification products were purified with Wizard® SV Gel and PCR Clean-Up System (Promega, Madison, WI, USA). Both strands were sequenced using the ABI Big Dye Terminator Cycle sequencing kit v 2.0 (Applied Biosystems, Foster City, CA, USA) on an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems). Sequences of CytB from all the S. subsignatus specimens collected from different geographical locations were subjected to multiple alignments using ClustalW (Thompson et al., Reference Thompson, Higgins and Gibson1994). Phylogenetic analysis was performed with PAUP*, version 4.0b4a (Swofford, Reference Swofford2003). A maximum-parsimony analysis was carried out on data set under a heuristic search strategy with all sites weighted equally, 1000 random addition sequences and TBR branch swapping. To assess the branching support, non-parametric bootstrapping was performed with 2000 replicates (Felsenstein, Reference Felsenstein1985). The CytB sequence of Curculio humeralis (Curculionidae: Curculioninae), GenBank accession number AY327729, was used as an outgroup taxon. The Tamura-Nei model of the Mega4 programme (Tamura et al., Reference Tamura, Dudley, Nei and Kumar2007) was used to calculate the distance (±SE) between sequences. The Tamura-Nei model was chosen because the average distance among SSW sequences was 0.051, 0.057 and 0.06 using the p-distance, Kimura-2-parameters and Tamura-Nei model, respectively. With distances in the range of 0.0–0.2, differences with the different methods are minimal (Nei & Kumar, Reference Nei and Kumar2000; Russo et al., Reference Russo, Miyaki, Pereira and Matioli2001); therefore, the method of choice is that which is the most simple and carries the least variance. Trees constructed with our sequences using p-distance, Kimura-2-parameter and Tamura-Nei are identical. The use of the Tamura-Nei model can also be justified by the transition/transversion ratio, which is 1.4 (the expected value is 0.5) and a bias toward AT (T=38.6%, C=16.5%, A=33.5%, G=11.4%). Strict consensus trees were created with the programme TREEVIEW, version 1.5.2 (Page, Reference Page2001).
Results
RAPD analysis
The size of this molecular marker varied between 300 bp and 2500 bp. This technique produced 236 polymorphic bands that were analyzed by the Dice coefficient and the UPGMA method (fig. 2). The Dice coefficient ranged between 0.13 and 0.91; the highest values were observed in samples 130 and 131 from São Desiderio. Sternechus populations clustered according to their geographical origin. Populations from Warta, Londrina Co. were more closely related genetically to some individuals from San Agustin population, Tucumán, Argentina, than the populations sampled in northeast Brazil. Specimens collected from Mauá da Serra, Paraná state, produced two discrete clusters, separated by gender, that were linked to specimens collected in northeast Brazil.

Fig. 2. Dendrogram constructed based on RAPD data of individual Sternechus subsignatusDNA template. The Dice coefficient and UPGMA method in NTSYS-pc software were used (Rohlf, Reference Rohlf1993). Values on the branches represent the percentages of individuals that fell into the group to the right (out of 1000 iterations).
SSW from Tasso Fragoso, Bahia state, diverged from the other populations. One cluster of this population grouped with the population collected in neighbouring localities of Sambaíba Co., Maranhão state, Formoso do Rio Preto and São Desidério, Bahia. Northeast Brazilian populations clustered in linked groups and were related.
Bootstrap analysis of the consensus tree indicated well-supported groups in the Warta population (82%), Formoso do Rio Preto (94%) and São Desiderio population (96%). Mauá da Serra, Sambaíba and Tasso Fragoso cluster nodes were lower than 80%, representing non-significant bootstrap analysis. San Agustin specimens grouped into two separate clusters with bootstrap values of 98% and 83% (fig. 2).
AMOVA results partitioned the genetic variation of Sternechus populations (table 2). A considerable proportion of the total genetic variation (29.1%) was observed among geographical populations and 70.9% was observed within populations. These results indicate that the SSW population is structured.
Table 2. Analysis of molecular variance among and within geographic populations of Sternechus subsignatus, based on RAPD data.
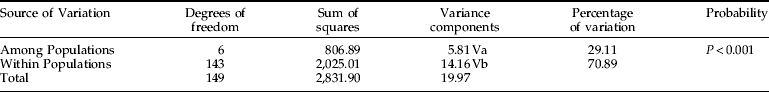
Va, variance among populations; Vb, variance within populations; fixation index ɸST=0.29.
Sequencing
The CytB fragments were ca. 470 bp. Sequencing results from the CytB fragment averaged 340 bp and the nucleotide sequences were deposited in the NCBI GenBank (Accession number DQ858208, DQ858209, DQ858210, EF141342, EF141343, EF141344, EF141345, EF141346, and EF141347). Of a total of 467 characters, 377 were conserved, 73 were variable and 17 were parsimony informative. The sequences were biased toward A and T nucleotides, as observed in a previous study (Simon et al., Reference Simon, Frati, Beckenbach, Crespi, Liu and Flook1994). Base composition averages were 34.05% A, 38.17% T, 16.69% C and 11.07% G. Differences between Argentinean and northeast Brazilian populations were responsible for most of the polymorphism (table 3). Comparisons among sequences (obtained from insects of different geographic origin using Parsimony analysis) suggested that most individuals from Tucumán, Argentina, were not divergent from Brazilian populations; a specimen collected in Mauá da Serra clustered with Argentinean populations of SSW (fig. 3). The mtDNA sequences of the longest amplicons (St-1 and St-10) corresponded to positions between 10,910 and 11,376 of the Drosophila melanogaster sequence (AF 00829.1), which encodes Cytb gene (Garesse, Reference Garesse1988).

Fig. 3. Strict consensus tree generated by parsimony analysis as implemented by Phylogenetic Analysis Using Parsimony (PAUP4.0) using the heuristic search algorithm. This is an arbitrarily chosen representative from 1000 equally parsimonious trees. Bootstrap analysis of 2000 replicates was performed using the Parsimony method. Numbers shown on the branches are bootstrap percentages derived from parsimony. Only values >50% are shown. Collection locations and dates are described in table 1. San Agustin represents the Sternechus subsignatus Argentinean population.
Table 3. Cytochrome B gene sequence divergence (lower-left side of the diagonal) and standard errors (upper-right side of the diagonal) between Sternechus subsignatus individuals based on pairwise comparisons (Tamura-Nei model).

Discussion
Sequencing and RAPD analysis of the mitochondrial cytB gene could not differentiate specimens collected in Argentina and Brazil. From an alignment of 213 sites, only 11 sites were divergent. Southern and northern populations could not be tested for reproductive compatibility because it is no possible to recreate the complete biological cycle of this species in the laboratory.
Clusters based on UPGMA and the RAPD Dice coefficient from the Warta and Mauá da Serra populations did not include specimens from other localities. The highest Dice coefficients of similarity were observed in the Formoso do Rio Preto and Warta populations, 0.67 and 0.60, respectively, suggestive of low differentiation potentially due to recent population of SSW in these areas (C. Berneche de Oliveira, personal observation). Populations of SSW caused economic losses in central-north and south Paraná during the 1983–1984 growing season (Oliveira & Hoffmann-Campo, Reference Oliveira and Hoffmann-Campo1984), whereas local populations from Warta began to increase during the 2001–2002 soybean-growing season (D. Sosa-Gomez, personal observation).
Populations from Sambaíba and Tasso Fragoso, BA, and Formoso do Rio Preto, MA, formed linked clusters, likely due to the geographic proximity and gene flow among these populations. No studies on flight behaviour of SSW have been published. However, SSW shows low mobility, dispersing no more than 23 m in soybean fields, at least following post-pupal emergence (Silva, Reference Silva1998). The most divergent and heterogeneous population was collected in San Agustin, Tucumán, Argentina. A group from this region was linked to Warta specimens, suggesting genetic heterogeneity (fig. 2).
Taxonomic studies based on morphological features provide evidence that the SSW that damages soybean fields in Argentina belongs to the species found throughout Brazil (G. Rosado-Neto, personal observation). Apparently, the early specimens, collected by Costilla in Tartagal, Salta, Argentina, that were identified by Charles W. O′Brien (Center for Biological Control Florida A&M University, Tallahassee, FL, USA) as S. pinguis (Costilla & Venditti, Reference Costilla and Venditti1990), actually belong to the S. subsignatus species, as do populations reported more recently in western and southern Tucumán Province and north of Santa Fe Province (Salas et al., Reference Salas, Medina, Casmuz and Antoni2002; Sosa, Reference Sosa2002).
Fast dispersing species recently introduced in Brazil, such as Bemisia tabaci, or clonally reproducing species, such as aphids, show little differentiation among populations (ca. 20.0%) (Lima et al., Reference Lima, Campos, Moretzsohn, Navia and de Oliveira2002; Lopes da Silva et al., Reference Lopes da Silva, Tonet and Vieira2004). In the case of S. subsignatus, the fixation index ɸST was 0.29, suggesting structured populations. RAPD analysis and fragments of the cytB gene sequence also showed similar patterns of genetic diversity among the populations; the Argentinean population grouped with specimens from Mauá da Serra with moderate bootstrap support (61%; fig. 3).
Soybean growers of northeast Brazil have assumed that population increases are a consequence of introductions or expansions of southern populations. RAPD and sequencing data, however, revealed that SSW outbreaks and population increases in northeast Brazil (west of Bahia and south of Maranhão states) are due to local genotypes. Indeed, south Brazil populations are not responsible for increases of the SSW populations in northeast Brazil.
Acknowledgements
The authors would like to thank Karina Lucas Silva Brandão, Carlos Caio Machado and Carlos A. Arrabal for reviewing the manuscript and Ana Claudia Barneche de Oliveira and Mauricio Conrado Meyer for collecting specimens in the Brazilian states of Bahia and Maranhão, respectively. This work was supported financially by CNPq (Conselho Nacional de Desenvolvimento Científico e Tecnológico, Edital Universal) and Embrapa Soja. This paper was approved for publication by the editorial board of Embrapa Soja as manuscript 018/2006.






