


Introduction
Phylogenetic analysis of DNA sequence data, in conjunction with associated approaches such as identification of species based on DNA sequences (barcoding), has facilitated both the identification of robust taxonomic groupings and also helped to clarify their systematic interrelationships (Besansky et al., Reference Besansky, Severson and Ferdig2003; Hajibabaei et al., Reference Hajibabaei, Singer, Hebert and Hickey2007). Such approaches have proven most useful in highly speciose taxa, especially where morphological similarity precludes identification of species using traditional cladistic approaches (Hebert et al., Reference Hebert, Penton, Burns, Janzen and Hallwachs2004; Smith et al., Reference Smith, Woodley, Janzen, Hallwachs and Hebert2006).
DNA-derived phylogenies have proven extremely useful in defining the biodiversity and systematics of midges of the genus Culicoides Latreille 1809. Culicoides midges are the major vectors of bluetongue virus (BTV), African horse sickness virus (AHSV) and epizootic hemorrhagic disease virus (EHDV) of the family Reoviridae. BTV infection can cause severe clinical disease, primarily in sheep, leading to high levels of mortality, with symptoms including oedema in the pharyngeal cavity and nasal discharge. Cattle and wild ruminants can develop a viraemia, but show fewer signs of disease. The recent incursion and spread of bluetongue disease in Europe has been linked to climate change, with warmer winters and higher precipitation providing conditions which promote the survival of midge vectors (Purse et al., Reference Purse, Mellor, Rogers, Samuel, Mertens and Baylis2005). The classical vector, Culicoides imicola, has been found in areas where it has not previously been present. Between 1998 and 2005, outbreaks of bluetongue affected 12 countries in the Mediterranean and central European regions about 800 km north of the expected geographic range. Moreover, expansion and propagation of BTV in these areas has been facilitated by endemic palaearctic species. Two widespread and abundant species groups are the Culicoides obsoletus sensu lato group and the Culicoides pulicaris sensu lato group. Members of both species groups have been shown to be able to transmit BTV (Carpenter et al., Reference Carpenter, Lunt, Arav, Venter and Mellor2006), and the overlapping geographical expansion of C. imicola and palaearctic species facilitated a crossover of the infection. In 2006, outbreaks of bluetongue serotype 8 were observed for the first time in The Netherlands, Belgium, Germany, France and Luxembourg (Meiswinkel et al., Reference Meiswinkel, van Rijn, Leijs and Goffredo2007; Wilson et al., Reference Wilson, Carpenter, Gloster and Mellor2007); and, in 2007, the first cases of BTV-8 were observed in southern England.
There are 30 Culicoides species involved in the transmission of orbiviral diseases. The internal taxonomy of the genus Culicoides has traditionally relied mainly on morphological identification based upon variation in wing pattern and male genitalia (Boorman, Reference Boorman1986). Therefore, in many cases, only males can be differentiated, even by specialists; and such traits are used without any recourse to potential phenotypic plasticity or geographic variation among individuals or populations. Moreover, this morphological variation cannot be used to gauge evolutionary distance and, thus, does not allow relative placement of the individual species in a broader Culicoides phylogeny (Edwards, Reference Edwards, Edwards, Oldroyd and Smart1939; Downes & Kettle, Reference Downes and Kettle1952; Khalaf, Reference Khalaf1954; Fox, Reference Fox1955). Application of DNA-based phylogenetic analysis has facilitated reliable species identification within the species complexes. Sequence analyses based on variations of the mitochondrial cytochrome oxidase I (COI) gene (Linton et al., Reference Linton, Mordue (Luntz), Cruickshank, Meiswinkel, Mellor and Dallas2002; Dallas et al., Reference Dallas, Cruickshank, Linton, Nolan, Patakakis, Braverman, Capela, Capela, Pena, Meiswinkel, Ortega, Baylis, Mellor and Mordue (Luntz)2003; Nolan et al., Reference Nolan, Carpenter, Barber, Mellor, Dallas, Mordue (Luntz) and Piertney2007) and the internal transcribed spacer regions I (ITSI) (Perrin et al., Reference Perrin, Cetre-Sossah, Mathieu, Baldet, Delecolle and Albina2006; Mathieu et al., Reference Mathieu, Perrin, Baldet, Delecolle, Albina and Cetre-Sossah2007) and II (ITSII) (Gomulski et al., Reference Gomulski, Meiswinkel, Delécolle, Goffredo and Gasperi2006) have been used to delimit species, primarily to develop diagnostic DNA-barcoding assays for the rapid and economical identification of individuals of unknown species provenance.
Less effort has been focussed on phylogenetic relationships among the species. In the context of BTV transmission, related species may express similar abilities to transmit the virus, which is the main motivation to resolve the exact phylogeny of the genus Culicoides. A major unresolved issue is the phylogenetic position of C. dewulfi, which is found from Britain to Eastern Europe. The importance of C. dewulfi as a bluetongue vector results from its breeding behaviour in cow dung. As a reservoir host for BTV, cattle can become viraemic without developing clinical signs of bluetongue disease characteristic of sheep, which facilitates the silent spread of the disease and also represents a possible overwintering mechanism. The role of C. dewulfi as a vector is further underlined by its affinity with species of the C. obsoletus s.l. complex. Insect trapping in central Europe revealed that the main vector for BTV in this climate zone is C. obsoletus due to dominant presence (Mehlhorn et al., Reference Mehlhorn, Walldorf, Klimpel, Jahn, Jaeger, Eschweiler, Hoffmann and Beer2007). C. dewulfi has, however, also been trapped in bluetongue-affected areas along with C. obsoletus sensu strictu and C. scoticus (Meiswinkel et al., Reference Meiswinkel, van Rijn, Leijs and Goffredo2007).
Several publications have discussed the phylogenetic position of C. dewulfi. It belongs to the subgenus Avaritia along with C. imicola and the C. obsoletus s.l. complex. Some consider it a member of the C. obsoletus s.l. complex (Savini et al., Reference Savini, Goffredo, Monaco, Di Gennaro, Cafiero, Baldi, de Santis, Meiswinkel and Caporale2005; Mathieu et al., Reference Mathieu, Perrin, Baldet, Delecolle, Albina and Cetre-Sossah2007; Nolan et al., Reference Nolan, Carpenter, Barber, Mellor, Dallas, Mordue (Luntz) and Piertney2007), while other sources have preferred to keep the species as a separate entity outwith the C. obsoletus s.l. complex (Conte et al., Reference Conte, Goffredo, Ippoliti and Meiswinkel2007; Meiswinkel et al., Reference Meiswinkel, van Rijn, Leijs and Goffredo2007). All phylogenetic studies so far have used only a small region of a single gene to resolve variation among Culicoides taxa, which precludes robust phylogenetic placement of C. dewulfi, and also introduces the potential that the phylogenies produced are actually gene trees and not true representations of species relationships (Gomulski et al., Reference Gomulski, Meiswinkel, Delécolle, Goffredo and Gasperi2006; Perrin et al., Reference Perrin, Cetre-Sossah, Mathieu, Baldet, Delecolle and Albina2006; Nolan et al., Reference Nolan, Carpenter, Barber, Mellor, Dallas, Mordue (Luntz) and Piertney2007). Here, we analyse the phylogenetic relationship among the Culicoides species, based on DNA sequence variation from three concatenated genetic markers, to produce a more robust phylogeny based upon combined nuclear and mitochondrial variation. We show that C. dewulfi indeed is a phylogenetically distinct clade positioned between the C. obsoletus s.l. complex and C. imicola.
Material and methods
Data mining
The majority of sequences were retrieved by database search. COI sequences were obtained for C. obsoletus (AM236652), C. scoticus (AM236625), C. dewulfi (AM236672), C. chiopterus (AM236751), C. pulicaris (AM236708), C. punctatus (AM236733), C. impunctatus (AM236717), C. newstaedi (AM236738), C. imicola (EU189056), and Aedes aegypti (DQ792578). ITSI sequences were obtained for C. obsoletus (AY861152), C. scoticus (AY861160), C. dewulfi (DQ408045), C. chiopterus (DQ408543), C. pulicaris (AY861156), C. punctatus (AY861157), C. impunctatus (AJ417986), C. newsteadi (AY861151), C. imicola (AY861144), and A. aegypti (M95126). ITSII sequences were obtained for C. obsoletus (AY599780), C. scoticus (AY599796), C. dewulfi (AY599818), C. pulicaris (DQ371264), C. punctatus (DQ371246), C. newsteadi (DQ371257), C. imicola (AY599832), and A. aegypti (M95126).
PCR
ITSII sequences for C. chiopterus (EU8206) and C. impunctatus (EU908207) were amplified by PCR using the primers 5′gatgaagaccgcagcaaact3′ and 5′atttgggggtagtcacacat3′ (Gomulski et al., Reference Gomulski, Meiswinkel, Delécolle, Goffredo and Gasperi2006) in a volume of 50 μl using the HotStarTaq Mastermix Kit (Qiagen, UK) with 0.4 μM of each primer and approximately 20 ng of DNA. Amplifications were done by an initial denaturation at 94°C for 15 min, then 32 cycles of denaturation at 94°C for 30 s, primer annealing at 55°C for 30 s, primer extension at 72°C for 1 min and, then, a final extension phase at 72°C for 6 min. PCR products were purified with QIAquick columns (QIAGEN, UK). Sequencing was performed at Eurofins MWG Operon (Martinsried, Germany).
COI, ITSI and ITSII sequences for each species were concatenated.
Phylogenetic analysis
The sequences were aligned in Bioedit version 5.0.9 (www.mbio.ncsu.edu/BioEdit). The phylogenetic relationships among taxa were resolved using a maximum likelihood approach in PAUP v4b10 (Swofford, Reference Swofford1998), with the resultant topology rooted using A. aegypti. The maximum likelihood method was preferred to other methods because it allows phylogenetic analysis based on probabilistic models of molecular evolution.
The program MODELTEST v3.06 (Posada & Crandall, Reference Posada and Crandall1998) was used to determine the most suitable model of DNA substitution. This analysis was done without the A. aegypti DNA sequence data included to optimise the phylogenetic relationship of Culicoides species. The model of evolution identified by the Akaike Information Criterion (AIC) had base frequencies of A=0.3132, C=0.1598, G=0.1730, T=0.3540; a proportion of invariable sites of 0; and a Gamma distribution shape parameter of 0.6538. The rate matrix parameters were A to C=1.4169, A to G=2.2331, A to T=1.6856, C to G=0.8305, C to T=3.4798 and T to G=1. These parameters were used in a heuristic search, and the reliability of the resulting tree topology was ascertained by bootstrapping (1000 replicates). Insertion and deletion mutations were removed from the analysis.
To test the hypothesis that C. dewulfi groups outside the C. obsoletus s.l. complex, the position of C. dewulfi was exchanged with other species within the phylogeny such that the number of different constrained trees in the file, each containing a different C. dewulfi position, equalled the number of species. Shimodaira-Hasegawa (SH) tests (Shimodaira & Hasegawa, Reference Shimodaira and Hasegawa1999) under full maximum likelihood optimisation were used to examine whether the optimal tree was significantly more likely than any of the constrained trees under the same model of sequence evolution. The SH test provides a likelihood-based statistical test of competing evolutionary hypotheses that have not been specified a priori, but instead when one of the topologies is the maximum likelihood topology for the dataset that was analysed; as such, the SH test details whether the resolved optimal topology is more likely than any topology constrained to satisfy an alternative hypothesis (Goldman et al., Reference Goldman, Anderson and Rodrigo2000).
Results
A maximum likelihood phylogenetic analysis, derived from an alignment of COI, ITSI and ITSII sequences of the nine Culicoides species and A. aegypti as an outgroup, resulted in a tree with clades that were well supported by bootstrapping (fig. 1). The species C. obsoletus, C. scoticus and C. chiopterus formed one clade. The species C. pulicaris, C. punctatus, C. impunctatus and C. newsteadi formed a second clade. C. dewulfi grouped well outside these two clades, as did C. imicola. The topology of the tree was consistent with those derived from maximum parsimony and distance-based neighbour-joining analyses (data not shown).

Fig. 1. Phylogenetic relationships among COI, ITSI and ITSII sequences of nine Culicoides species and the outgroup species Aedes aegypti, based on a maximum likelihood analysis. The values at the nodes indicate the percentage of times the clade to the right was retained when the tree was redrawn (1000 times) using randomly sampled data with replacement from the original dataset (bootstrapping).
Removal of the A. aegypti outgroup did not change the topology of the C. obsoletus and C. pulicaris complexes nor their high level of bootstrap support as reciprocally monophyletic clades. However, C. dewulfi grouped with C. imicola, and there was a strong bootstrap support for this shared clade (fig. 2).

Fig. 2. Phylogenetic tree based on a maximum likelihood analysis using COI, ITSI and ITSII sequences of nine Culicoides species without an outgroup species.
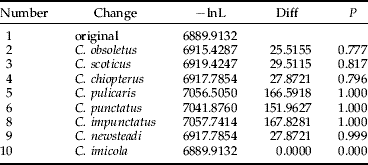
The SH analysis clearly indicated that the resolved maximum likelihood tree was significantly more likely than any other topology where C. dewulfi was placed elsewhere (table 1).
Table 1. Relative maximum likelihood scores for resolved phylogeny and phylogenies constrained to place C. dewulfi elsewhere within the topology. Probability values, P, that the constrained topology is less likely as the resolved topology are provided (Shimodaira-Hasegawa test).
Discussion
This study has highlighted that C. dewulfi should not be considered part of the C. obsoletus s.l. complex, and instead should be defined as a separate grouping more closely related to C. imicola. Previous phylogenetic analyses have placed C. dewulfi close to the C. obsoletus s.l. complex, but these were based either upon single gene sequences (Nolan et al., Reference Nolan, Carpenter, Barber, Mellor, Dallas, Mordue (Luntz) and Piertney2007) or were compromised by including too few species (Gomulski et al., Reference Gomulski, Meiswinkel, Delécolle, Goffredo and Gasperi2006; Mathieu et al., Reference Mathieu, Perrin, Baldet, Delecolle, Albina and Cetre-Sossah2007). A species tree, resulting from the analysis of several genes combined, is expected to yield a more accurate phylogeny than single gene trees, which can be biased by different mutation rates, recombination events, effective population sizes, etc. Moreover, as phylogenetic analysis of closely related species should include independent sequences with high rates of mutation (Slade et al., Reference Slade, Moritz and Heideman1994), our approach, combining a mitochondrial gene and two nuclear sequences in a single analysis, should produce a robust phylogeny.
The so-called species-complexes of the genus Culicoides are essentially arbitrary groupings based upon the similarity of constituent species in wing morphology and male genitalia. C. dewulfi was placed in the C. obsoletus s.l. complex because it most closely resembles C. chiopterus in these traits. C. dewulfi has a similar geographic distribution to members of the C. obsoletus s.l. complex, as evidenced from light-trapping in bluetongue-affected areas (Savini et al., Reference Savini, Goffredo, Monaco, Di Gennaro, Cafiero, Baldi, de Santis, Meiswinkel and Caporale2005; Meiswinkel et al., Reference Meiswinkel, van Rijn, Leijs and Goffredo2007). It also breeds in unadulterated dung heaps, as does C. chiopterus (Kettle, Reference Kettle1977). It is clear, however, that morphological and behavioural similarities do not reflect evolutionary history and genetic similarity; and, as such, there are no grounds for grouping C. dewulfi with C. chiopterus in the C. obsoletus s.l. complex.
Based on our data, we would consider C. chiopterus to be part of the obsoletus complex as our analyses group C. obsoletus and C. scoticus in a well-supported clade. If C. obsoletus sensu strictu and C. scoticus are usually considered as one complex (Mellor & Wittmann, Reference Mellor and Wittmann2002), then DNA sequence analysis strongly supports C. chiopterus to be a member of this complex. Much less controversy has occurred about the so called C. pulicaris s.l. complex, which includes five species as indicated in a study by Nolan et al. (Reference Nolan, Carpenter, Barber, Mellor, Dallas, Mordue (Luntz) and Piertney2007), and this complex is well bootstrap-supported by our results.
The phylogeny of the tree indicates that, during evolution, C. imicola branched off from a common ancestor first, followed by C. dewulfi and then the ancestors of the two complexes. If the level of vector-competence is a feature of closely related species, then the close relationship of C. dewulfi with the classical Old World vector C. imicola recommends a close monitoring of potential hosts in areas at risk of bluetongue outbreaks where this species is abundant. However, the different grouping of vector-competent species suggests that this feature has evolved on more than one occasion. This highlights (i) the need for the vector competence of all Culicoides species to be ascertained and (ii) that the assessment of disease risk for BTV should not be based upon the presence of C. obsoletus s.l. group vectors but, potentially, all Culicoides midge species.
Acknowledgements
This work was funded by DEFRA grant SE4101. We thank our colleagues at the Institute for Animal Health in Pirbright, UK, for sending specimens of adult midges.



