


Introduction
In classical biological control, collection of natural enemies in the area of origin, quarantine procedures, and bulking for release present opportunities for genetic changes in the population. For successful establishment, an introduced species needs rapid adjustment to the new environmental conditions. Selection constraints may favour adaptable genotypes and so could modify the genetic make-up during co-selection for genes conferring fitness under these new conditions. The effect of these events on eventual adaptability of the resultant population is unclear (Omwega & Overholt, Reference Omwega and Overholt1996; Hufbauer, Reference Hufbauer2002; Hufbauer et al., Reference Hufbauer, Bogdanowicz and Harrison2004; Lloyd et al., Reference Lloyd, Hufbauer, Jackson, Nissen and Norton2005; Zayed et al., Reference Zayed, Constantin and Packer2007) and the lasting genetic signal depends on the size of the founding population (Grevstad, Reference Grevstad1999; Sakai et al., Reference Sakai, Allendorf, Holt, Lodge, Molofsky, Kimberly, Baughman, Cabin, Cohen, Ellstrand, McCauley, O'Neil, Parker, Thompson and Weller2001; Memmott et al., Reference Memmott, Craze, Harman, Syrett and Fowler2005). Understanding the genetic changes occurring during these phases is an essential aspect of auditing a biological control activity.
The expansion of the ecological range of a species subjects it to ecological and genetic constraints that affect establishment success. In classical biological control and species invasion, introductions may generate an initial genetic exclusion reducing allelic diversity (Nei et al., Reference Nei, Maruyama and Chakraborty1975). Although large introductions are recommended in biological control, the size of the founder population alone is not informative without knowledge of the underlying diversity, which may be exploited to increase the variability of the populations to be released in biological control programs (Lockwood et al., Reference Lockwood, Cassey and Blackburn2005; Bacigalupe, Reference Bacigalupe2008). Diversity can also be reduced in laboratory culturing if strong selection forces exist (Bigler, Reference Bigler and Ochieng-Odero1992). Despite the importance of the genetic parameters, little work has been done on auditing the biological control process through the genetics of natural enemies.
The larger grain borer (LGB) Prostephanus truncatus (Horn) (Coleoptera: Bostrichidae) is the most serious post-harvest pest of maize in Africa (Schneider et al., Reference Schneider, Borgemeister, Setamou, Affognon, Bell, Zweigert, Poehling and Schulthess2004). It has been reported in 21 countries spreading from sites of initial introduction in Togo and Tanzania some 30 years ago (Schneider et al., Reference Schneider, Borgemeister, Setamou, Affognon, Bell, Zweigert, Poehling and Schulthess2004; Gueye et al., Reference Gueye, Goergen, Badiane, Hell and Lamboni2008; Nyagwaya et al., Reference Nyagwaya, Mvumi and Saunyama2010). Two populations of Teretrius nigrescens Lewis (Coleoptera: Histeridae) collected from Mexico and Costa Rica (some 2200 km apart) were released in East and West Africa, respectively (Böye, Reference Böye, Markham and Herren1990; Giles et al., Reference Giles, Hill, Nang'ayo, Farrell and Kibata1996; Hill et al., Reference Hill, Nang'ayo and Wright2003; Schneider et al., Reference Schneider, Borgemeister, Setamou, Affognon, Bell, Zweigert, Poehling and Schulthess2004). The establishment of T. nigrescens and resultant LGB control varied with the population introduced and the agroecological conditions in the area of release (Schneider et al., Reference Schneider, Borgemeister, Setamou, Affognon, Bell, Zweigert, Poehling and Schulthess2004; Omondi et al., Reference Omondi, Jiang, van den Berg and Schulthess2011a ). While the Costa Rica population reportedly successfully established and effected control in Benin, Togo, Eastern Ghana and Nigeria, the Mexican population established only in Kenya, where the pest incidence has resurged. Although the West African releases are thought to be more successful than the East African ones, no comparative studies exist between them. Biochemical, morphometric and ecological differences between populations of the LGB from Mexico, Togo and Costa Rica have been reported (Guntrip et al., Reference Guntrip, Silby and Smith1996; Mendiola-Olaya et al., Reference Mendiola-Olaya, Valencia-Jimenéz, Valdés-Rodríguez, Délano-Frier and Blanco-Labra2000) but little is known of T. nigrescens diversity, nor the genetic diversity of the LGB.
The native ecological range of the two species spans around 4000 km – from southern USA to Ecuador – covering a wide range of agroecological conditions. We hypothesized that genetic adaptability attributes might exist and that they contributed to the efficiency of T. nigrescens to control LGB in Africa. In an earlier study, we had shown the existence of two parapatric mitochondrial COI lineages of T. nigrescens populations (Omondi et al., Reference Omondi, van den Berg, Masiga and Schulthess2011b ). To gain further insight into the genetic differentiation, gene flow and demographic history of the biological control agent we developed microsatellite markers and used them to study the population structure and demographic history of the predator. The availability of independent isolations, and information on management and release strategies of T. nigrescens offers a unique opportunity to study the genetic aspects of the differences in its establishment and efficacy. We therefore investigated the genetic structure of local populations of T. nigrescens in Central America and those released in Africa. We tested the hypothesis that two populations introduced in Africa were distinct genetic races, and that isolation, culturing, release and establishment of T. nigrescens left genetic constraints. Thus we compared the neutral genetic variation of 13 populations of T. nigrescens spanning a geographical transect in central America, different introduction histories in biological control, field sources, and rearing regimes as well as recoveries from established populations. To determine if culturing left genetic signatures in the populations, we compared sample populations recovered from the field in regions where recoveries from biological control were done with samples released in Africa; and daughter colonies with parents from which they had been subcultured. We also compared African releases with field populations from Central America. Knowledge of the genetic structure of T. nigrescens populations is important in setting genetic standards when prospecting for new populations, monitoring released populations and their establishment.
Materials and methods
Insects and sample preparation
Teretrius nigrescens populations were sampled from Central America and Africa as summarized in table 1, representing ca. 2200 km of the native range from Central Mexico to Costa Rica (fig. 1). Alcohol-preserved samples from three populations from Ghana and Malawi were kind donations from Dr Richard Hodges, NRI, UK and Dr Roderick Ndawala (NARO, Lilongwe), respectively. The Mombasa population was sampled using Delta sticky flight traps (Pherocon II, Trécé, Salinas, USA) baited with LGB aggregation pheromone lure. All other field recoveries were done using funnel traps baited with synthetic LGB pheromone vials, LGB frass and maize as described in Omondi et al. (Reference Omondi, Jiang, van den Berg and Schulthess2011a ).

Fig. 1. Field origins of the populations of T. nigrescens used in this study. 1- El Batan, 2- Tlaltizaspan, 3- Oaxaca, 4- Nuevo Leone (Kenyan releases), 5: Guanacaste, Costa Rica (West African releases), 6-Gualaso, 7-Yoro, 8-Teupasenti. Shaded circles represent field sources of the six populations recovered from Africa. Sample 4 gave rise to KARI, Kiboko and perhaps Store; whereas sample 5 gave rise to Benin, NRI and Malawi.
Table 1. Field sources and culturing history of the sample populations of T. nigrescens used in this study.

1 Ethanol-preserved samples, only used for molecular studies. The size of the original field-recovered population of the CIMMYT samples was about 200 beetles. The original size number of traps used and sampling duration for the recovery of KARI and Benin populations are not known.
2 T. nigrescens from Benin was also released in Tanzania from where they could have dispersed into Kenya. Samples from the field in Kenya were therefore treated as possibly from both ancestries until after COI typing described in (Omondi et al., Reference Omondi, van den Berg, Masiga and Schulthess2011b ).
DNA extraction and genotyping
Genomic DNA was extracted from the head and thorax of individual T. nigrescens adults, using the phenol–chloroform–isoamyl alcohol protocol (Sambrook et al., Reference Sambrook, Fritsch and Maniatis1989) with slight modifications (cf. Omondi et al., Reference Omondi, van den Berg, Masiga and Schulthess2011b ). Genotyping was done using 16 microsatellite markers, with fluorescent dye-labelled forward primers and unlabelled reverse primers. DNA from 492 individuals from 13 populations was genotyped as described in Omondi et al. (Reference Omondi, Orantes, van den Berg, Masiga and Schulthess2009). Allele sizing was done in an ABI sequencer, with LIZ 500 internal standard and scored using Genemapper Software version 7 (ABI Biosciences) at the BeCA Hub, ILRI, Nairobi, Kenya.
Data analysis
Genetic diversity
Microchecker software (van Oosterhout et al., Reference van Oosterhout, Hutchinson, Willis and Shipley2004) was used to evaluate the existence of null alleles for each locus. Genepop version 3.4 (Raymond & Rouset, Reference Raymond and Rouset1995) was employed to test for population departure from HWE, and linkage disequilibrium using Markov Chain Monte Carlo (MCMC) methods with 10,000 replications and 10,000 dememorization steps. The allelic distribution and variation were determined using FSTAT version 2.9.3.2 (Goudet, Reference Goudet2002) and GenAlex 6.5 (Peakall & Smouse, Reference Peakall and Smouse2006, Reference Peakall and Smouse2012). Hierarchical analysis of molecular variance (AMOVA) was done to partition of the variation observed among populations and groups and its significance tested with 999 permutations, using GenAlex 6.5. Principle Coordinate analysis (PCoA) was run to further assess the population clustering of the populations based on three principal coordinates from pairwise distances between populations and between individuals.
Demographic history
Recent demographic events such as migration, population bottlenecks and expansions leave a genetic signature in the genotypes and allele frequencies in (Cornuet & Luikart, Reference Cornuet and Luikart1996; Luikart et al., Reference Luikart, Allendorf, Cornuet and Sherwin1998). To test the hypothesis of population size constancy, two indicators were examined. First, each population was analysed for heterozygote deficiency or excess using Bottleneck version 1.2 (Luikart & Cornuet, Reference Luikart and Cornuet1998; Piry et al., Reference Piry, Luikart and Cornuet1999) with 10,000 iterations. The Two-Phase Model (TPM) assuming up to a maximum of 50% of the infinite allele mutation model (IAM) was used with 10,000 replications. Demographic decisions were based on Wilcoxon's test for the probability of deviation from HWE. Secondly, the populations were tested for genetic signatures of expansion by analysing the deviation from mutation–drift equilibrium using the intra-locus k-test and inter-locus g-test (Reich et al., Reference Reich, Feldman and Goldstein1999) in Kgtests (Bilgin, Reference Bilgin2007). The k statistic tests whether the frequency distribution of allele lengths is more peaked than would be expected for a population of constant size, a sign of expansion. The g statistic tests for the presence of lower variance among loci in the variance of allele frequency sizes than expected for a constant sized population. The results were compared with the fifth percentile cut-off values (depending on the number of loci and sample size) (Reich et al., Reference Reich, Feldman and Goldstein1999).
In populations with subtle structure, allele frequency tests may be less sensitive, yet bottlenecks, expansions and founder effects may be tested by examining the proportion and frequency of rare alleles relative to a reference field population (Hundertmark & van Daele, Reference Hundertmark and van Daele2010). We therefore tested the hypothesis that population fragmentation (as through sub-culturing or introduction) would lead to a greater loss of rare alleles in subpopulations, and that founder effect alters the allele frequencies. The density and frequency of: private alleles, rare alleles (P<5%; P<25% and P<50% across populations) and alleles shared by just two of the 12 populations, was computed and compared.
Population structure analysis
FSTAT version 2.9.3.2 was used to calculate the genetic differentiation (FST) and to test its significance within and between populations. Structure version 2.2 (Pritchard et al., Reference Pritchard, Stephens and Donnely2000; Falush et al., Reference Falush, Stephens and Pritchard2003, Reference Falush, Stephens and Pritchard2007) was applied to test for the existence of a genetic structure between and within geographic populations and to visualize individual membership to the inferred populations. We used a panmictic genetic mixture model to infer the number of genetic populations (K) and likelihood of assignment of individuals to each population.
To estimate the number of genetic populations, two coalescent modelling approaches were used: two ad hoc (Evanno et al., Reference Evanno, Regnaut and Goudet2005; Pritchard et al., Reference Pritchard, Wen and Falush2007) and one learning approach (Beaumont et al., Reference Beaumont, Barratt, Gottelli, Kitchener, Daniels, Pritchard and Brudford2001) for each K (2–20) the analysis was run in four replicates using an admixture ancestry model, with migration prior of 0.05 and initial value for alpha inference=1 (Pritchard et al., Reference Pritchard, Wen and Falush2007). A different lambda was inferred for each population from the genotype data. Burn-in period was set at 10,000 and running length to 100,000; null alleles coded as recessive alleles (Falush et al., Reference Falush, Stephens and Pritchard2007). The likely true number of populations was inferred by plotting probability of K [Ln P(D)] against values of assumed K between 1 and 20, predicting the true value of K as the point where the curve enters the plateaus phase (fig. 1). The approach of Evanno et al. (2005) was used on these results to determine the most likely K, and to compare with the result obtained (Pritchard et al., Reference Pritchard, Wen and Falush2007). In the third approach, the reference population method (PopFlag=1 option) of Beaumont et al. (Reference Beaumont, Barratt, Gottelli, Kitchener, Daniels, Pritchard and Brudford2001) was used. Finally, a population assignment test was done in GenAlex 6.5 using both the strict ‘leave one out’ and the less conservative ‘assign all’ settings, respectively. The proportion of the populations assigned within their own population and region was compared.
Isolation by distance
To test the hypothesis that geographical distance influenced the patterns of differentiation observed, the association between pairwise genetic distances (FST/1−FST) and orthodromic geographic distances (km) between locations of field origin of the populations was done using the web-based IBDWS Version 3.15 software (Jensen et al., Reference Jensen, Bohonak and Kelley2005). This analysis was done separately for samples from Central America and those from Africa. Mantel (Reference Mantel1967) test was employed with 30,000 randomizations.
Results
Genetic diversity
All loci were polymorphic in at least one population, with a mean of 3.8 alleles per locus (table 2; Supplementary Table 1). Private alleles were more common in the field populations than in African introductions, while their frequency showed the opposite trends. Similarly, putative parent populations had a higher proportion but lower frequency of private alleles per population.
Table 2. Genetic variability indicators calculated from allele frequencies of 16 polymorphic microsatellite loci of T. nigrescens populations from three regions.

nA, average number of alleles; nR±SD, allelic richness and Standard deviation; HE, mean expected heterozygosity, HO, mean observed heterozygosity, Hs, gene diversity index and standard deviation, An, number of null alleles; FIS, coefficient of inbreeding, (* shows that values are significantly different from zero [p<0.05]), np, number of private alleles, Ap, mean frequency of private alleles.
Most populations did not deviate significantly from the HWE expectations for most alleles. As analysis using Microchecker detected a significant signal of null alleles in five loci for the Teupasenti population we used 16 loci (of the 21 originally developed) for this study (Omondi et al., Reference Omondi, Orantes, van den Berg, Masiga and Schulthess2009). The field populations sampled from Central America were all more genetically diverse than populations released in Africa for Biological control (KARI and Benin). Within respective African releases, all daughter populations were less diverse than their parent laboratory colonies (KARI>Store/Kiboko; Benin>Malawi/Ghana). Also, field samples had a larger number of private and rare alleles than laboratory colonies. Within colonies, pairwise FIS was high for field populations but generally lower for daughter populations.
Genetic differentiation
The observed pairwise genetic distances (FST) reflect the geographical origin of the populations. Between groups of populations, higher pairwise genetic distances were observed between the southern populations (Honduras/Costa Rica populations) and Mexico populations than between populations within the two major divisions (table 3). From each group, populations were more closely related to those from the same region of origin. Curiously, the Oaxaca population showed closer similarity with the Benin population, sampled from Costa Rica three decades prior, than to the simultaneously sampled, geographically proximate Mexican populations. The highest pairwise genetic differences are between populations caught in Kenya with all other populations. Within itself, the Kenyan release also represents the lowest genetic identity within its group (table 3). These could be both a result of genetic isolation forced by culturing practices and geographical distance and genetic drift effects in small colonies.
Table 3. Genetic differentiation (pairwise FST) between populations based on 16 neutral markers (below diagonal) and the number of shared rare alleles 1 (above diagonal).

1 Rare alleles are those that were shared between just two populations and exclude private alleles. The distribution of private alleles in populations is given in table 2. Numbers in bold are significant at 5% adjusted for multiple comparisons.
Teretrius nigrescens was genetically structured within populations, between populations within a region and between regions (table 4). Variation between individuals within populations was the main contributor to the genetic diversity observed, accounting for 71% of the variance. Variations between regions and between populations within a region were also highly significant contributing to 19 and 10% of the observed variation, respectively.
Table 4. AMOVA for 12 populations assuming three population groupings based on geographical origin and release for biological control in Africa.
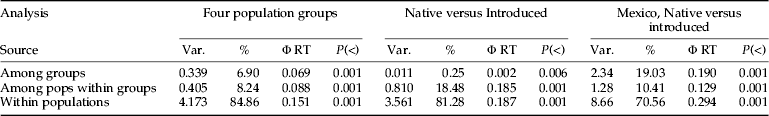
Var, Variance estimate.
Analysis was done for all four population groups as used in this manuscript: Mexico (Batan, Oaxaca and Tlaltizaspan): Ex-Mexico (KARI, Kiboko and Store), Costa Rica (Benin, Malawi and Ghana) and Honduras (Yoro, Teupasenti and Gualaso). Second analysis redistributed populations between Native (Mexoco and Honduras) versus introduced (Costa Rica group and Ex-Mexico group): Finally the second analysis was repeated only with Mexico and Ex-Mexico groups.
The most likely number of genetic populations was estimated at five (figs 2 and 3). At this level, all samples from Mexico, Honduras and Kenya occupied each respective distinct clusters (fig. 4). Samples from Costa Rica were split between two remaining clusters, with Benin in one cluster and its daughter populations Ghana and Malawi occupying a different cluster. Insects from Oaxaca were allocated to the same clusters as Benin (49.1%); Mexico (36%) and Kenya (13%). This population represents an admixture of genotypes at this level but clusters with Mexican populations at lower levels of assumed K (Supplementary Figure S1).

Fig. 2. Estimation of the population structure of T. nigrescens based on the gradient of assumed number of populations (K) against log likelihood of [L(K)].

Fig. 3. Detection of the number of clusters of individuals based on the rate of change in log-likelikood of K (ΔK) (Evanno et al., Reference Evanno, Regnaut and Goudet2005). The asterisk marks the inferred K. Sharp changes in gradient after K=5 suggest the existence of genetic substructure.

Fig. 4. Results of cluster analysis using Bayesian methods based on 16 neutral microsatellite loci. The graphical representation of the data set for the most likely number of genetic clusters (K=5), each colour representing a suggested cluster and each individual is represented by a single vertical bar. Values on the y-axis represent the probability of a genotyped individual's membership to one of the five colour-coded inferred genetic clusters: red – Mexico group (Batan, Tlaltizaspan); Green – Costa Rica Group (Benin); Yellow – Malawi/Ghana subpopulations; Blue – KARI Group (KARI, Store, Kiboko); and Pink – Honduras group (Teupasenti, Gualaso, Yoro). The table below the distruct plot shows the mean proportion of individual membership to each cluster.
Significant peaks at K=8 and 11, depict a genetic substructure within the main clusters (Supplementary material Fig. S1). Indeed stepwise allocation of population to individual clusters always followed a geographic signal. The PCoA with both the population matrix (fig. 5) and individual similarity matrix (Supplementary material Fig. S2) showed a similar result. The contribution of each principle coordinate to the total separation is shown on each respective figure, but was higher for population PCoA than pairwise individual PCoA. The first dimension separated the populations originating from Mexico from those from Honduras and Costa Rica together (similar to the COI separation Omondi et al., Reference Omondi, van den Berg, Masiga and Schulthess2011b and Structure K=2 Supplementary Fig. 1). The second dimension separated the Benin samples from the Honduras samples, but did not resolve populations of Mexican origin. All samples grouped by their original geographical origin at least in the first two dimensions. In population assignment prediction, at least 54% of the time, individuals were assigned back to their sampling populations, and almost always to their proper groups (table 5; Supplementary Table 2). No individual from the African samples (Benin, Malawi, Ghana) and (KARI, Kiboko, Store) are assigned to the others group (Costa Rica and Ex-Mexico, respectively). Interestingly, Ex-Mexico populations released in Kenya are assigned to this unique class but less to Mexico region.

Fig. 5. Two-dimensional scaling on three first principle coordinates calculated from population pairwise FST values.
Table 5. Proportion of population assignment using microsatellite pairwise differences for individual samples.

1 Assignment of individuals to these populations is derived as a sum to their assignment to populations making a single group sensu table 2. Figures in bold face show the probability of assignment to their source populations with the conservative ‘leave last out’ setting.
Demographic history
Allele frequency distributions did not detect bottlenecks except for the Gualaso population. With the intralocus k-test, the Benin, KARI, Malawi, Yoro and Gualaso populations did not show a population expansion signature (table 6). When populations were compounded by region to test the possible effect of population structure on this result, all clusters showed a significant population expansion signal. However, the interlocus g-test, which is less sensitive to population size or genetic substructure, remained positive indicating that the hypothesis of population size constancy could not be rejected. However, analysis of rare alleles shows a loss of diversity between Central American populations, African recoveries and their daughter populations. The test for rare alleles is not affected by substructure and so could be most reliable in this situation (Hundertmark & van Daele, Reference Hundertmark and van Daele2010). These data demonstrate the loss of diversity and drift effects in culture.
Table 6. Intragenomic (k) and intergenomic (g) tests for population expansion of T. nigrescens populations.

1 Populations were compounded based on geographical proximity of the areas of origin. Recent Mexico populations (Batan, Oaxaca and Tlaltizaspan); All Mexico populations (recent Mexico populations, Field, KARI and Store populations); Costa Rica populations (Benin, Malawi and Ghana).
* p≤0.05.
Geographical differentiation
Genetic distance was significantly positively correlated with geographical distance in South American samples but not for African samples (n=28; Z=4062.2459; r=0.711, P=0.0058) (fig. 6) and (Z=8147.4030, r=0.031, P=0.347), respectively. Similar results are obtained when the African samples are used and geographical locations taken as those of original field recovery in Central America.

Fig. 6. Graph of RMA regression for pairwise geographic orthodromic distances (km) and pairwise genetic distances [FST/(1−FST)] between meso-American populations.
Discussion
This study shows that T. nigrescens is genetically structured in its native range in America. The populations released in Africa for the control of the LGB have maintained their similarity with geographical populations in their area of origin. As shown previously (Omondi et al., Reference Omondi, van den Berg, Masiga and Schulthess2011b ), they also remained separated from each other since independent introduction into Africa three decades ago. The data also show rapid drift following subculturing, releases or establishment events, and associated loss of rare alleles. The populations released in Africa represented only a small part of the T. nigrescens gene pool, which could restrict their ability to spread and colonize new environments.
Five genetic populations were observed with the Oaxaca samples showing a mixed ancestry, consistent with previous results that reported two lineages sympatric in Oaxaca (Omondi et al., Reference Omondi, van den Berg, Masiga and Schulthess2011b ). Although this earlier study suggested the existence of phylogenetically distinct races, hierarchical substructure in the present study showed genetic variation with geographical distance. Genetic diversity was significantly higher in field than laboratory populations, and in parent laboratory cultures than in daughter sample populations (e.g., Benin>Malawi or Ghana; KARI>Kiboko, Store and Mombasa). Although allele frequency-based tests for bottlenecks and expansion were negative, a direct measure based on the proportion and frequency of rare alleles showed a clear reduction of the former and increase of the latter. A lower occurrence of private alleles in African releases shows that sampling or colony establishment excluded some genotypes relative to the parent population while a higher frequency of rare alleles depicts founder effect (Kalinowski et al., Reference Kalinowski, Taper and Marshall2007; Szpiech and Rosenberg, 2011). As populations subdivide each fragmentation step is likely to result in inadequate sampling of the genetic diversity of the population while increasing the relative frequency of sampled rare alleles in the new population. Loss of allelic diversity may lead to the overexpression of deleterious genes or depletion of rare but important fitness linked alleles. These might in turn result in increased sensitivity to environmental fluctuations or pathogens, thereby reducing the potential establishment and success of released individuals.
Significant signals of genetic changes in culture, or during recovery or establishment events may explain the differences between field recovered samples and their parent cultures, or between parent and daughter subcultures. While Store and Kiboko were both divergent from each other and from the parent population (see PCA plot), the Malawi and Ghana samples were more closely related to each other but not as closely to their parent population. The Benin and Ghana samples had a recent history of introgression with wild individuals recaptured in Africa (Meikle et al., Reference Meikle, Rees and Markham2002). These observations suggest changes in allele frequencies due to genetic drift, sampling effects or colony subdivision, and stochastic effects of genetic changes associated with prolonged laboratory culturing (Lloyd et al., Reference Lloyd, Hufbauer, Jackson, Nissen and Norton2005). These results underline the importance of increasing the genetic diversity of new introductions of natural enemies to enhance the chances of establishment.
The level of ecological differences associated with these findings is unclear. The data presented here show that T. nigrescens sorted into two large clusters, and within them, genetic populations. All populations were most similar to those from their native area of origin. However, the Oaxaca population presents a possible transition zone or point of origin of the predator from where dispersal occurred in opposite directions. The existence of ecotypes of the predator has been speculated before as a possible explanation for the inconsistent performance of the predator in West and East Africa (R. Hodges, NRI, UK; personal communication). However, the predator had also been released in much higher numbers and more sites in West than in East Africa (Giles et al., Reference Giles, Hill, Nang'ayo, Farrell and Kibata1996). A greater propagule size and larger release area should have favoured the establishment of a predator (Grevstad, Reference Grevstad1999), especially those relying on prey-emitted kairomones like T. nigrescens. One would also expect diversity to be directly correlated to the resilience of T. nigrescens in a new environment (Gomulkiewicz et al., Reference Gomulkiewicz, Holt, Barfield and Nuismer2010).
Anecdotal reference to the existence of ecotypes of the pest P. truncatus has been made (Guntrip et al., Reference Guntrip, Silby and Smith1996; Vasquez-Arista et al., Reference Vasquez-Arista, Smith, Martinez-Gallardo and Blanco-Labra1999) but genetic evidence is lacking. It is suggested that the ability of the predator in maintaining an association with its prey in the new environment as in its native range would depend on matching ecotypes of the two (Omwega & Overholt, Reference Omwega and Overholt1996; Lloyd et al., 2003; Roderick & Navajas, Reference Roderick and Navajas2003). A comparison of the adaptability of the putative geographical populations to different ecological conditions would verify whether there are true ecological races. Such a comparison should take into consideration weather patterns, agrocological characteristics, predator fitness and predator–prey genetic matching. This information should help to design a release strategy and improve the probability of establishment of the predator in a given ecozone.
Genetic diversity of natural enemies is a key parameter in biological control. It determines the eventual establishment, ecological fitness and effectiveness of introduced natural enemies, because diversity provides a pool of genes on which selection occurs (Roush, Reference Roush, Baker and Dunn1990; Roderick & Navajas, Reference Roderick and Navajas2003). Rapid adaptation to local environmental conditions and eventual effectiveness depends on the suitability of the species introduced, and the existence of well-adapted geographical biotypes. However, the process of collection, quarantine rearing and importation promotes the maintenance of small populations which may impose a series of bottlenecks and favour a reduction of genetic diversity (Hopper & Roush, Reference Hopper and Roush1993; Hopper et al., Reference Hopper, Roush and Powell1993; Lloyd et al., Reference Lloyd, Hufbauer, Jackson, Nissen and Norton2005; Fauvergue et al., Reference Fauvergue, Malausa, Giuge and Courchamp2007). Furthermore, logistical limitations and sheer scarcity in the field dictate that natural enemies are often collected as small subpopulations, often ill-representing the full genetic diversity in the source population (Lloyd et al., Reference Lloyd, Hufbauer, Jackson, Nissen and Norton2005), and loss of potentially important rare alleles may occur (Nei et al., Reference Nei, Maruyama and Chakraborty1975; Bigler, Reference Bigler and Ochieng-Odero1992). The differences between African populations and their daughter samples support this observation. The populations of T. nigrescens released in Africa have been separated from the mother populations for about 30 years now (Giles et al., Reference Giles, Hill, Nang'ayo, Farrell and Kibata1996; Meikle et al., Reference Meikle, Rees and Markham2002) corresponding to just 400 generations of the predator. This is too short in species evolution to allow for significant mutations.
That the genetic structure of T. nigrescens was significantly related to its geographic distribution has key implications in biological control. An understanding of possible topographical barriers to gene flow in the native range would help design an effective recovery–release plan. As ecotypes of the LGB have been reported (Tigar et al., 1994; Guntrip et al., 1994), the two populations of the predator recovered from Central America and eventually released in Africa represent a very narrow coverage of the gene pool. We still do not know which ecotype(s) of the LGB invaded Africa. Reasonably, either the native associated populations of T. nigrescens or a wide diversity of the predator should be considered for releases in Africa. Large ecologically diverse field isolations and releases may boost the genetic diversity of starter colonies, increasing the chance that fitness-relevant genes are harnessed.
The association between neutral genetic changes and fitness-related traits is debatable (van Tienderen et al., Reference van Tienderen, de Haan, van der Linden and Vosman2002; Bekessy et al., Reference Bekessy, Ennos, Burgman, Newton and Ades2003) and should be investigated directly using sequence tagged markers. Colonies of T. nigrescens were founded by 200 individuals and the smallest releases were done with 5000 individuals (Giles et al., Reference Giles, Hill, Nang'ayo, Farrell and Kibata1996; Anonymous, 1999; P. Likhayo, KARI, Nairobi; I. Hoeschle-Zeledon, IITA, Ibadan, Nigeria, personal communication). Teretrius nigrescens has shown inconsistent establishment and control success in Africa, prompting some workers to conclude that the natural enemy is unsuitable for biological control (Holst & Meikle, Reference Holst and Meikle2003). Yet, a reduction in damage, LGB flight activity and grain loss was reported following the release of the predator (Hill et al., Reference Hill, Nang'ayo and Wright2003; Schneider et al., Reference Schneider, Borgemeister, Setamou, Affognon, Bell, Zweigert, Poehling and Schulthess2004). The reduction reported in the former study was later attributed to changes weather patterns (Omondi et al., Reference Omondi, Jiang, van den Berg and Schulthess2011a ), consistent with the resurgence of the LGB in previous control regions in Kenya. As LGB remains an important post-harvest pest in Africa, genetically diverse releases may improve the establishment and success of T. nigrescens. These findings, therefore, open an opportunity to increase adaptability by genetic re-introgression with fresh field and established populations between regions in Africa and with Mesoamerica, and monitoring of the specific released local populations of the predator. A detailed genetic study of the pest would also help identifying the original invasive population(s) and potentially isolate adapted populations of T. nigrescens.
The supplementary materials for this article can be found at http://www.journals.cambridge.org/ber
Acknowledgements
The Teretrius nigrescens samples used in this study were kind donations from the following colleagues: Richard Hodges (NRI, Greenwich, UK), Paddy Likhayo and Josephine Songa (KARI, Kiboko, Kenya), Rhoderick Ndawala (NARO, Lilongwe, Malawi), Cypriene Atcha (IITA, Abomey Calavi, Benin) and David Bergvinson (CIMMYT, Mexico). We thank Lucia Orantes and Alfredo Rueda (Zamorano University, Tegucigalpa, Honduras) for field sampling in Honduras. We express sincere appreciation to Sarine Adhiambo (ICIPE) and Robert Mutweti (KARI, Katumani, Kenya) for technical assistance. We also thank Drs Dr Pascal Campagne (IRD), Miriam Karlsson, Dirk-Louis Schorkopf (SLU, Sweden); Joana Ferreira Marques, Franklin Nyabuga (Lund University, Sweden) for useful comments on various versions of the manuscript. This study was supported by the Kirkhouse Trust, UK, through a PhD Fellowship to BOA. Field sampling in Honduras was funded by the Swiss Development Cooperation. Genotyping work was accomplished through a Fellowship at Biosciences East and Central Africa (BeCA-Hub), ILRI, Nairobi.













