


Introduction
Pine wilt disease caused by the pine wood nematode Bursaphelenchus xylophilus (Steiner & Buhrer) is the most serious forest disease in Japan and continues to expand its distribution northwards. One of the most important issues in protection against pine wilt disease is stopping its range expansion as it continues to move northwards. The pine sawyer Monochamus alternatus (Hope) (Coleoptera: Cerambycidae) is the main vector of the pine wood nematode in Asian countries (Mamiya & Enda, Reference Mamiya and Enda1972). Therefore, knowledge of the dispersal of the pine sawyer is essential when developing an intensive protection programme against pine wilt disease.
Damage arising from pine wilt disease can be seen as far as the Tohoku district on the northeastern part of Honshu Island. The Ohu mountain range runs north to south throughout this area. Damage arising from pine wilt disease has progressed northwards synchronously on both sides of the mountain range (Kamata, Reference Kamata1996). Since it first invaded Tohoku in 1975, the disease has moved approximately 100 km northwards on the east side of the mountain range and 200 km on the west side (fig. 1).
Movement of the sawyer population can be inferred by tracing the dispersal of pine wilt disease. The disease does not occur at high altitudes (Kishi, Reference Kishi1995), indicating that the mountain range acts as a geographical barrier to sawyer migration. Because the adult sawyer can feed on other conifers as well as Pinus species (Nakamura & Okochi, Reference Nakamura and Okochi2002), it is thought to be able to live in various types of forests. It might, therefore, be possible for the sawyers to migrate between mountains.
Within Akita Prefecture, western area of the frontier populations, there appears to be two dispersal directions at a smaller spatial scale. One from the south, where the disease was introduced in 1982, and the other from the Oga Peninsula, where a sudden invasion occurred in 1988 (fig. 1). Due to the absence of a nearby damaged forest, the 1988 invasion is likely to have originated from a timber yard at the port containing logs that had been infested with both vectors and nematodes. The damaged area has expanded from the south and from Oga, and the pine wilt disease from each direction seems to have met.
Recently, methods using genetic data have been employed to estimate the dispersal dynamics of organisms. Traditionally, Wright's F-statistics have been used (e.g. Lehmann et al., Reference Lehmann, Hawley, Kamau, Fontenille, Simard and Collins1996); however, this method assumes that a population is in demographic equilibrium, which is not the case in many natural populations (Bohonak, Reference Bohonak1999). Regional equilibrium refers to a condition whereby a balance between the loss of alleles due to drift and their replacement by gene flow occurs between populations within a region (Hutchison & Templeton, Reference Hutchison and Templeton1999). Non-equilibrium situations arise from historical factors such as range expansion, recent bottlenecks, and secondary contact of populations (Nei et al., Reference Nei, Maruyama and Chakraborty1975; Turgeon & Bernatchez, Reference Turgeon and Bernatchez2001).
Several methods that do not assume an equilibrium population have been used in recent studies (e.g. Noor et al., Reference Noor, Pascual and Smith2000; Manel et al., Reference Manel, Gaggiotti and Waples2005). For example, the development of an assignment test and its applications has influenced quantitative studies on the dispersal of individuals (e.g. Berry et al., Reference Berry, Mandy, Tocher and Sarre2004). Multidimensional scaling, which is a class of ordination techniques that displays the complex relationships between populations in a small number of dimensions (Lessa, Reference Lessa1990), also provides a method for examining dispersal in nonequilibrium populations. This approach uses genetic distance to reveal major barriers to dispersal in a given study area. Multidimensional scaling can also show the relative degree of differentiation between populations. Finally, interest is increasing in using microsatellite DNA techniques to assess dispersal in populations that are not in equilibrium (Rousset, Reference Rousset, Colbert, Danchin, Dhondt and Nichols2001; Sved et al., Reference Sved, Yu, Dominiak and Gilchrist2003; Berry et al., Reference Berry, Mandy, Tocher and Sarre2004).
Genetic differentiation of M. alternatus has been found amongst prefectures throughout Japan (Kawai et al., Reference Kawai, Shoda-Kagaya, Maehara, Zhou, Lian, Iwata, Yamane and Hogetsu2006). No information, however, is available about genetic structure at local spatial scales in Tohoku. Microsatellite DNA analysis has potential to show the local spatial genetic characteristics of frontier populations and their genetic dynamics, such as the dispersal of individuals within a region, as well as stochastic processes by which frontier populations are established.
The present study had three goals. The first was to use microsatellite markers to map the current population structure of M. alternatus near the frontier of the pine wilt disease invasion. Second, these data were used to examine the dispersal of the M. alternatus populations and individuals. The final goal was to relate changes at local and regional scales to changes in gene flow and associated stochastic processes.
Materials and methods
Sample collection
Monochamus alternatus emerges from the trunks of pine trees, and disperses from late June to September in Tohoku district, Japan. The sawyers in this area are univoltine or semivoltine. Sawyers were collected in 2003 and 2004 from nine sites in Tohoku district: six sites were on the western side of Ohu in Akita Prefecture (A1–A6) and three were on the eastern side in Iwate Prefecture (I1–I3), which is near the frontier of the pine wilt disease invasion (fig. 1). Within each prefecture, the geomorphology is flat and no obvious geographical barriers exist. Adult sawyers were collected weekly from July to September using attraction traps at each site except at A3 and A4, where adults were sampled as they emerged from logs; 9–41 individuals were collected per site (table 1). All samples were preserved in 99.5% ethanol at 4°C.

Fig. 1. Sampling locations and spread of pine wilt disease in Tohoku district, Japan from 1975 to 2004.
Table 1. Number of alleles across samples (N a), observed (H O) and expected (H E) heterozygosities and departures from Hardy–Weinberg equilibrium (F IS) for all samples and loci.

Likelihood ratio test: NS, not significant; * P<0.05; ** P<0.01.
F IS based on Weir & Cockerham (Reference Weir and Cockerham1984).
A1–A6, samples of Monochamus alternatus from Akita Prefecture; II–I3, samples of M. alternatus from Iwate Prefecture.
DNA extraction and microsatellite genotyping
DNA was extracted using the Chelex method (De Lamballerie et al., Reference De Lamballerie, Zandott, Vignoli, Bollet and de Micco1992; Honda et al., Reference Honda, Nakashima, Yanase, Kawarabata and Hirose1998) with modifications as described by Kawai et al. (Reference Kawai, Shoda-Kagaya, Maehara, Zhou, Lian, Iwata, Yamane and Hogetsu2006). The following five microsatellite loci were examined: Mal A, Mal B, Mal F, Mal G, and Mal I (T. Hogetsu, T. Maehara & Z. Zhou, unpublished data). Polymerase chain reaction (PCR) was performed in 8-μl volumes containing one tenth volume 10× PCR buffer with Mg2+ (TaKaRa), 160 μm each dNTP, 0.4 units of Taq polymerase (TaKaRa), 40 ng of fluorescently labelled primer, 40 ng of correspondence tailed primer (Applied Biosystems), and genomic DNA. Amplifications were as follows: four cycles at 94°C for 30 s, 47°C for 30 s, and 72°C for 45 s; followed by six cycles at 94°C for 30 s, 50°C for 30 s, and 72°C for 45 s; then 20 cycles at 94°C for 30 s, 53°C for 30 s, and 72°C for 45 s. For multiplex loading, 0.5 μl of each PCR product and 0.5 μl of GeneScan 400HD [ROX] Standard (Applied Biosystems) were added to 12 μl of deionized formamide. Products were separated using capillary electrophoresis (ABI PRISM 310; Applied Biosystems) and assigned scores using 310 GeneScan software (Applied Biosystems).
Data analysis
Fundamental genetic parameters were calculated for all loci using POPGENE version 1.31 (Yeh et al., Reference Yeh, Yang and Boyle1999). Observed (H O) and expected heterozygosities (H E) were calculated to quantify the genetic diversity of each population. To test for bottlenecks, we used the computer program BOTTLENECK program version 1.2 (Cornuet & Luikart, Reference Cornuet and Luikart1996) and a two-tailed Wilcoxon's signed-rank test to look for the deficiency or excess of heterozygosity under the infinite-allele-model (IAM). FSTAT version 2.9.3 (Goudet, Reference Goudet1995) was used to test for the deviation from linkage disequilibrium at each locus using the Markov chain method. F ST and F IS (smallif) (Weir & Cockerham, Reference Weir and Cockerham1984) were calculated with FSTAT to test for local inbreeding within populations and differentiation between populations. The significance of population differentiation was tested assuming Hardy–Weinberg equilibria within populations by the permutation test. The post hoc test was adjusted by sequential Bonferroni correction (Rice, Reference Rice1989).
Multidimensional scaling based on Nei's (Reference Nei1978) unbiased genetic distances D was used to explore the relationship between geographical sites and genetic differentiation. Multidimensional scaling is a procedure for fitting a set of points in a space such that the distances between points correspond as closely as possible to a given set of dissimilarities between a set of objects. This was performed using the SYSTAT program, version 9.01 (SPSS Inc., 1998). The fit of the data in two dimensions was measured by the stress factor. A hierarchical analysis of molecular variance (nested AMOVA) was calculated using ARLEQUIN 2001 (Schneider et al., Reference Schneider, Roessli and Excoffier2000) to divide the microsatellite variance into the following components: geographical locations across the mountain range (between Akita and Iwate prefectures), locations within the prefectures but among populations (A1–I3), and locations within populations (i.e. among individuals). A1–A6 and I1–I3 were nested into Akita and Iwate, respectively. By using an AMOVA, we were able to use F ST to quantify the degree of population differentiation. The significance of any differentiation was tested using the permutation method with 10,000 replications.
Isolation by distance testing can be used to infer the history and population structure in non-equilibrium populations (Hutchison & Templeton, Reference Hutchison and Templeton1999). lsolation by distance was assessed by testing the correlation between genetic and geographical distance using population pairs to estimate the regression of F ST/(1−F ST) on logarithm of distance for populations, as suggested by Rousset (Reference Rousset1997). This test was done amongst Akita Prefecture populations (A1–A6). The relationship between genetic differentiation (F ST/(1−F ST)) and geographic distance separating the populations (log metres) was assessed using the Mantel test (Mantel, Reference Mantel1967), with 9999 randomizations.
The proportion of each individual's genotype that originated from one of the other populations was estimated using the assignment tests. Frequency assignments using likelihood (Paetkau et al., Reference Paetkau, Calvert, Stirling and Strobeck1995) and partial Bayesian assignments (Rannala & Mountain, Reference Rannala and Mountain1997) were calculated with GENECLASS 2 (Piry et al., Reference Piry, Alapetite, Cornuet, Paetkau, Baudouin and Estoup2004). The assignment threshold scores were 0.05. The accuracy of the assignment test was calculated as the percentage of sawyers that were correctly assigned to each site or prefecture.
Results
Genetic diversity and genotypic linkage
A total of 188 individuals was genotyped for each microsatellite locus. The number of alleles ranged from two (Mal G) to nine (Mal I); see table 1. The mean observed heterozygosity for all loci varied from 0.282 to 0.480 across the nine sites, with an overall mean of 0.364. Mal I had the highest expected heterozygosity (H E: 0.519–0.732) of the five loci, and Mal A had the lowest (H E: 0–0.296). Wilcoxon's signed-ranks test showed that none of the populations went through a significant bottleneck (P>0.05). The null hypothesis of Hardy–Weinberg equilibrium was rejected for the A3 population (P<0.01), but the multi-locus F IS values did not deviate significantly from zero for the other populations. Genotypic disequilibrium was not detected among populations (P>0.05) after sequential Bonferroni correction for multiple comparisons.
Population differentiations and assignment tests
The estimate of population differentiation (F ST) for all populations was 0.025, which was significantly positive (P<0.001). For the pair-wise comparisons, significant differentiation was only detected between the Akita and Iwate populations (table 2). The final configuration of the multidimensional scaling analysis showed a stress of 0.0547, explaining 98.2% of the variance (fig. 2). Along dimension 1, two major clusters could be found, A1–A6 and I1–I3. The AMOVA explained approximately 95% of the variability within sites (table 3). The remaining variation could be explained by the significant genetic heterogeneity between Iwate and Akita prefectures; however, variation amongst sites within prefectures was not significant. A significant pattern of isolation by distance was detected between sites within the Akita Prefecture populations based on the Mantel test (Mantel's r=0.477, P=0.0251; fig. 3).

Fig. 2. Multidimensional scaling analysis of Monochamus alternatus of frontier populations in Tohoku district, Japan based on Nei's (Reference Nei1978) unbiased genetic distances D.

Fig. 3. The relationship between genetic differences [F ST/(1−F ST)] and geographical distances in Monochamus alternatus within Akita Prefecture populations.
Table 2. Pairwise F ST values.
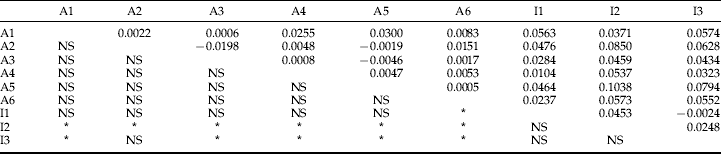
After sequential Bonferroni correction: NS, not significant; * P<0.05.
A1–A6, samples of Monochamus alternatus from Akita Preferture; II–I3, samples of M. alternatus from Iwate Preferture.
Table 3. Nested-AMOVA results for all individuals from the Akita and Iwate populations of Monochamus alternatus.
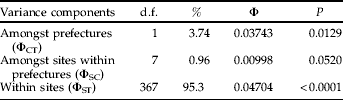
The percentage column indicates the amount of total variance explained by each hierarchical spatial scale. P-values were obtained by comparing observed values with those generated by random permutation. Φ-statistics are analogous to Wright's F-statistics.
The simple frequency and Bayesian likelihood assignments did not provide accurate assessments of genotype origin. Sawyers were correctly assigned to sites in 19.5–72.7% and 23.3–72.7% of cases for the frequency and Bayesian tests respectively (fig. 4); however, misassignment into populations for Iwate Prefecture was less than 25% for both methods.

Fig. 4. The percentage of Monochamus alternatus individuals correctly assigned to their sample sites and prefectures using the frequency assignment (Paetkau et al., Reference Paetkau, Calvert, Stirling and Strobeck1995) and partially Bayesian methods (Rannala & Mountain, Reference Rannala and Mountain1997). ■, frequency assignment;  , Bayesian assignment.
, Bayesian assignment.
Discussion
Genetic differentiation was only significant between Akita and Iwate prefecture sites and the multidimensional scaling revealed two clear groupings: A1–A6 and I1–I3. This supports the hypothesis that the Ohu mountain range acts as a geographical barrier. The large genetic distance between Akita and Iwate must therefore be a consequence of different colonizing routes because M. alternatus adults are unlikely to be able to migrate over the mountain range. Pair-wise F ST values were relatively low (−0.0198 to 0.0300) for populations in Akita Prefecture (A1–A6), and no significant differentiation occurred between populations. Gene flow within the Akita populations is thought to be high. It is possible that this species of sawyer beetle frequently settles in new pine forests with patchy distributions, and that it often moves between populations when no geographical barrier prevents dispersal.
The analysis also detected significant isolation by distance within the Akita populations. Such clinal genetic variation suggests two possibilities for adults dispersal. The first is that secondary contact took place as two invading populations expanded separately from the Oga Peninsula and from south Akita. The second is that the restricted gene flow corresponds to a stepping-stone model, which assumes that a finite set of populations is distributed in a regular lattice of patches, and dispersal takes place between neighbouring patches. The first possibility involves non-equilibrium change, whereas the second is an equilibrium situation. Hutchison & Templeton (Reference Hutchison and Templeton1999) described methods for discrimination between non-equilibrium and equilibrium conditions, but did not consider secondary contact. It is difficult to distinguish between the two possibilities using genetic information alone; however, the sawyer's colonization of this area is thought to be recent, suggesting that insufficient time has passed for the establishment of regional equilibrium. Because no significant bottleneck had occurred in the populations, the founder effects due to sequential colonization from the south or Oga might not have worked for these populations to form isolation by distance patterns. Therefore, the isolation by distance found within Akita Prefecture may be a result of secondary contact.
The accuracy of the assignment tests was poor for both the frequency method and Bayesian method, which was mainly due to the relative lack of specific alleles compared to dominant alleles for each locus. Therefore, it is difficult to predict the most likely source of origin of the populations, or to distinguish between immigrants and residents using the available microsatellite loci. To improve techniques for measuring dispersal of M. alternatus, it is necessary to develop additional microsatellite markers that are highly polymorphic, and therefore more informative.
The dispersal ability of the adult pine sawyer has been studied using mark and recapture methods and by measuring the rate of dispersal of pine wilt disease (Kamata, Reference Kamata1996). These studies revealed that most adult dispersal takes place over short distance increments (Shibata et al., Reference Shibata, Kawasaki and Takeda1986; Fujioka, Reference Fujioka1993) and that the dispersal distance of an adult throughout its life is 50–260 m (Togashi, Reference Togashi1989). The longest measured dispersal of a marked adult captured by an attractant trap using α-pinene and ethanol, which are volatiles from pine wood, was 2 km (Fujioka, Reference Fujioka1993). Dispersal distance using marked and recaptured individuals tends be underestimated because long-distance dispersal is ignored and these models include unconfirmed postulates (Roff & Yoccoz, Reference Roff, Yoccoz, Hanski and Gilpin2001). Therefore, complementary studies are important in evaluating the true dispersal distances.
Based on pine wilt disease tracking, the largest dispersal of an adult sawyer was estimated to be 2–20 km (Kamata, Reference Kamata1996). Propagation of pine wilt disease, however, can also be caused by human transportation of infested wood. Isolation by distance was detected even in the Akita Prefecture populations, indicating that individuals do not freely disperse between populations within a 28-km radius. Although it is difficult to infer the average or maximum dispersal distance from an assignment test, the populations throughout Akita Prefecture do not exhibit panmixia; that is, they are not randomly mating. Therefore, from a genetic standpoint, the dispersal distance of 20 km appears to be an overestimate.
The F IS value for all loci did not deviate significantly from 0 for all populations except A3. This is similar to other Japanese populations studied by Kawai et al. (Reference Kawai, Shoda-Kagaya, Maehara, Zhou, Lian, Iwata, Yamane and Hogetsu2006); that is, most sawyers mate randomly within sites. Because the A3 population was not collected using an attractant trap, the emergent adults were obtained from a relatively small sample of timber. This may mean the collected samples did not provide an accurate representation of the actual population, or that they were inbred. Furthermore, if the collection area of infested woods was too large to be panmictic, the Wahland effect may have occurred.
A process linking genetic structure at local (kilometres) and regional spatial scales (hundreds of kilometres) can be proposed based on the results of this study. Kawai et al. (Reference Kawai, Shoda-Kagaya, Maehara, Zhou, Lian, Iwata, Yamane and Hogetsu2006) found differentiation between prefectures and no isolation by distance throughout Japan, suggesting that the invasion of the pinewood nematode disturbed the genetic structure of M. alternatus populations. No information, however, is available on gene flow between sawyer populations or its stochastic mechanisms. On a local scale, adults frequently migrate to nearby forests, but long-distance dispersal is uncommon and adults rarely disperse over geographical barriers. They occasionally colonize distant habitats carrying pine wood nematodes. The resulting tree mortality creates abundant breeding material, thereby allowing the population of pine sawyer to increase rapidly. Throughout this process, stochastic effects are thought to be strong enough to cause significant differences in F ST values between prefectures. Within a prefecture, however, gene flow acts to effectively homogenise the genetic composition of populations. It is predicted that the populations studied in Akita Prefecture will eventually lose their isolation by distance and therefore become completely integrated. Continuous monitoring of genetic structure for several generations will provide further insight to the genetic dynamics of the metapopulation within this region.
Acknowledgements
The author thanks K. Fujita for cooperation in this study and comments on the manuscript and K. Kobayashi, K. Hoshizaki, T. Goto, K. Nakamura-Matori, H. Kinuura, A. Nagaki and K. Takahashi for valuable input and for setting up traps. H. Fujioka, T. Koiwa and J. Kon assisted in collecting the sawyers and T. Maehara, T. Hogetsu and Z. Zhou kindly allowed the use of the primer sets.
