


Introduction
Orthoptera is an order of insects with paurometabolous or incomplete metamorphosis, including more than 25,700 extant species of grasshoppers, crickets and katydids, many of which are notorious as pests worldwide. Orthopteran insects have diversified into various lineages that occupy every conceivable terrestrial habitat except the polar regions and play important roles in global ecosystems (Uvarov, Reference Uvarov1966; Kevan, Reference Kevan and Parker1982). Such diversity in form and function has attracted researchers who use these insects as models for studying bioacoustics, anatomy, physiology, chemical ecology, neurobiology, life-history traits, evolutionary ecology and speciation research (Uvarov, Reference Uvarov1966; Baccetti, Reference Baccetti1987; Chapman & Joern, Reference Chapman and Joern1990; Gangwere et al., Reference Gangwere, Muralirangan and Muralirangan1997; Pener & Simpson, Reference Pener and Simpson2009). The monophyly of Orthoptera is strongly supported by certain morphological characteristics, such as the presence of cryptopleuron and jumping hind legs (Kevan, Reference Kevan and Parker1982; Kristensen, Reference Kristensen1991; Grimaldi & Engel, Reference Grimaldi and Engel2005), and the order is considered to be divided into two suborders: Ensifera (crickets, katydids, wetas and their relatives) and Caelifera (grasshoppers, locusts and their relatives). Despite the familiarity and diversity of the group, the phylogenetic relationships within Orthoptera are poorly understood, and its higher classification remains unstable because of a long history of conflicting taxonomic hypotheses based on different character sets, fossil wing venation (Zeuner, Reference Zeuner1942; Sharov, Reference Sharov1968; Gorochov, Reference Gorochov1995a), internal organs (Slifer, Reference Slifer1939; Judd, Reference Judd1947; Baccetti, Reference Baccetti1987), external morphology (Blackith & Blackith, Reference Blackith and Blackith1968; Vickery & Kevan, Reference Vickery and Kevan1983) and the male phallic complex (Chopard, Reference Chopard1920; Ander, Reference Ander1939; Roberts, Reference Roberts1941; Dirsh, Reference Dirsh1973; Amégénato, Reference Amedegnato1974; Eades, Reference Eades2000). Most of these hypotheses lack formal analyses, and there are disagreements among hypotheses based on the same set of morphological characteristics. Several conflicting hypotheses on the phylogenetic position of the Orthoptera have been proposed, but they have not been well settled by experimental evidence (Hall, Reference Hall1999).
Currently, mitochondrial genomes are becoming increasingly common in higher-level phylogenetic analyses because of their ability to provide better resolution for deep relationships than single or multi-gene analyses and the feasibility of sequencing the entire genome with relative ease (Saccone et al., Reference Saccone, De Giorgi, Gissi, Pesole and Reyes1999; Chintauan-Marquier et al., Reference Chintauan-Marquier, Legendre, Hugel, Robillard, Grandcolas, Nel, Zuccon and Desutter-Grandcolas2016). Mitochondrial genome analyses may provide useful information for controlling insect pests. More abundant data indicate more reliable phylogenetic results; therefore, the sequencing of additional mitochondrial genomes might help to resolve the accurate phylogenetic relationships within Orthoptera. We first successfully amplified, sequenced and assembled the mitochondrial genome of Orthopteran species. The grasshopper C. fasciata (Ceracris, Oedipodinae, Acrididae, Orthoptera) studied in this paper is a major pest species, and it is mainly distributed in Yunnan, Guangdong, Guangxi, Hainan, Jiulong and Fujian Provinces in China. This species is harmful to bamboo shoots, Dendrocalamus brandisii, Bambusa lapidea, whangee and cotton bamboo. In addition, little is known about the biological characteristics and ecology of the DNA and the mitochondrial genome of C. fasciata.
Many new mitochondrial genomes of Orthopteran species are being added to Orthoptera; thus, the phylogenetic relationships of Orthoptera should be re-examined.
Correction or weighting of DNA sequence data based on the level of variability can improve phylogenetic reconstructions in certain cases. Therefore, gene choice is very important because rRNA genes are most likely to be useful at the population level and at deep levels of divergence (Zhang et al., Reference Zhang, Huang, Lin, Wang and Zheng2013a, Reference Zhang, Zeng, Huang and Zhengb). Protein-coding genes may be more appropriate for phylogenetic analyses at intermediate levels of divergence. The phylogenetic performance of different genes is related to their particular rates of evolution. The three protein-coding genes ATP6, ATP8 and NAD4L are the fastest evolving genes, while COX subunits and CYTB show much slower overall rates of evolution (Russo et al., Reference Russo, Takezaki and Nei1996; Zardoya & Meyer, Reference Zardoya and Meyer1996; Cameron et al., Reference Cameron, Miller, D'Haese, Whiting and Barker2004). COX1 is the most conserved gene in terms of amino acid evolution. In the past, COX1, COX2, CYTB and NAD2 were extensively used for phylogenetic analyses (Liu & Beckenbach, Reference Liu and Beckenbach1992; Simon et al., Reference Simon, Rati, Beckenbach, Crespi, Liu and Flook1994; Caterino et al., Reference Caterino, Cho and Sperling2000; Chapco et al., Reference Chapco, Litzenberger and Kuperus2001; Litzenberger & Chapco, Reference Litzenberger and Chapco2001; Chapco & Litzenberger, Reference Chapco and Litzenberger2002; Amédégnato et al., Reference Amédégnato, Chapco and Litzenberger2003). COX2 is the most widely used mitochondrial protein-coding gene in insects (Simon et al., Reference Simon, Rati, Beckenbach, Crespi, Liu and Flook1994). Both NAD4 and NAD5 belong to large genes, and their protein sequence divergences seem to be helpful in constructing trees for distantly related species (Russo et al., Reference Russo, Takezaki and Nei1996). The NAD6 gene has frequently been omitted because it is coded on the light strand, and its properties differ from those of the other 12 protein-coding genes (Springer et al., Reference Springer, DeBry, Douady, Amrine, Madsen, de Jong and Stanhope2001). Zardoya & Meyer (Reference Zardoya and Meyer1996) classified mitochondrial protein-coding genes into three groups: good (NAD4, NAD5, NAD2, CYTB and COX1), medium (COX2, COX3, NADl and NAD6) and poor (ATP6, NAD3, ATP8 and NAD4L) phylogenetic performers. Thus, certain genes seem to be consistently more reliable tracers of evolutionary history than others.
In this study, we established a robust phylogeny of Orthoptera, including 26 of 40 families representing 13 currently recognized superfamilies (according to the international classification system Orthoptera Species File) based on all codon positions of 13 protein-coding genes in mitochondrial genomes. Eighty-one complete mitochondrial genomes of Orthoptera were used in this text (table 1). The mitochondrial genome of C. fasciata not only built a more convincing analysis of phylogenetic trees by establishing phylogenetic relationships within Orthoptera but also provided more information for research into the prevention and control of this pest. Using our phylogeny, we tested previous phylogenetic hypotheses and propose a new classification scheme for the group. Using strongly corroborated fossil evidence, we present divergent time estimates for major Orthopteran lineages and also explore whether different lineages have undergone different tempos of diversification throughout 300 My of evolution.
Table 1. List of Orthoptera taxa used in this study.

Methods and materials
A sample of C. fasciata was collected in Baise, Guangxi, China in 2007. The specimens chosen in this paper were initially determined by Fauna Sinica and then received a final personal identification and verification by professor Guofang Jiang.
The C. fasciata sample was stored in 100% ethanol and maintained at −20°C. The total genomic DNA was extracted from the muscle of the specimen's femora using the standard proteinase K and genomic DNA purification kit (Promega, Beijing, China) according to the manufacturer's instructions (Sambrook & Russell, Reference Sambrook and Russell2001). Certain pairs of polymera chain reaction (PCR) universal primers and specific primers were designed to amplify overlapping segments of the entire mitochondrial genome of C. fasciata (see online table S1). Normal PCRs were performed in a 25 µl reaction mixture consisting of 16.3 µl of sterilized water, 2 µl MgCl2 5 mM, 2.5 µl 10 × PCR buffer, 2 µl dNTP at 2.5 mM, 0.5 µl of each primer at 10 µM, 1 µl DNA template (25–50 ng) and 0.2 µl Takara Taq DNA polymerase. Initial denaturation was performed for 5 min at 94°C, followed by 35 cycles of 30 s at 94°C, 30 s at 52–60°C, and 60 s at 72°C, and a subsequent 7 min final extension step at 72°C. The fragments (≥1 kb) were amplified using Takara LATaq (Takara Biomedical, Japan) under the following conditions: 5 min at 94°C, followed by 35 cycles of 30 s at 94°C (with 20 s added to each subsequent cycle), 30 s at 50–60°C, 1–2 min at 72°C and an extension period of 10 min at 72°C. The other fragments (<1 kb) were amplified using Takara rTaq (Takara Biomedical, Japan) under the following conditions: 5 min at 94°C, followed by 35 cycles of 30 s at 94°C, 30 s at 50–60°C, 20–60 s at 72°C and a final extension period of 10 min at 72°C. Then, all the PCR products were sequenced directly by Jin Si Rui Biotechnology Company, and the control-region was sequenced by Sangon Biotechnology Company.
Sequence data were assembled using SeqMan software (DNAStar, Inc.). Transfer RNAs were identified by tRNAscan-SE 1.21 (Storozhenko, Reference Storozhenko, Gangwere, Muralirangan and Muralirangan1997), and the other genes were determined through the comparison of sequences with previously published Ceracris mitochondrial genomes. Strand skew values were calculated according to the formulae by Perna & Kocher (Reference Perna and Kocher1995).
A total of 81 polyneopteran mitochondrial genomes available in GenBank were included in our analyses (table 1). For the outgroup, we excluded the taxa that had been reported to introduce phylogenetic errors based on the following criteria (Van Raay & Crease, Reference Van Raay and Crease1994): extreme compositional bias, inversion or translocation of genes on the opposite strand, inversion of the A + T-rich region, or the lack of ATP8. As a result, we chose 11 species from Gryllidae, Gryllotalpidae, Rhaphidophoridae, Prophalangopsidae, Anostostomatidae, Myrmecophilidae and Tettigoniidae. This selection appears appropriate considering both the insect phylogeny and outgroup choice in previous studies (Flook & Rowell, Reference Flook and Rowell1998; Terry & Whiting, Reference Terry and Whiting2005; Ma et al., Reference Ma, Liu, Yang and Kang*2009). We systematically surveyed a number of different data partitioning schemes according to previous studies (Leavitt et al., Reference Leavitt, Hiatt, Whiting and Song2013). We selected a scheme with 13 partitions. Partitioning by genes resulted in 13 partitions and 13 protein-coding genes. Annotated mitochondrial genomes were organized, with each gene aligned separately. We aligned the genes based on the conservation of reading frames by first translating them into amino acids, aligning them individually using default parameters and then click DNA Sequence button for back-translating from amino acids to nucleotides in MEGA 6.06 (http://www.megasoftware.net/) (Tamura et al., Reference Tamura, Stecher, Peterson, Filipski and Kumaret2013) followed by manual refinements. The maximum-likelihood (ML) and Bayesian analysis (BI) methods were used to reconstruct the trees. The concatenated dataset was partitioned by gene for the ML and BI searches. For the ML analyses, we used the best-fit partitioning scheme recommended by Partition Finder with the GTRGAMMA model applied to each partition and analysed trees using RAXML Blackbox implemented on the supercomputer resources available at the website (http://embnet.vital-it.ch/raxml-bb/). For the BI analyses, according to the MrModelTest program, the Akaike information criterion was used to select GTR + I + G as the best fit model for the sequence evolution for each gene partition. For the Bayesian phylogenies in MrBayes version 3.2 (Ronquist et al., Reference Ronquist, Teslenko, Van der Mark, Ayres, Darling, H€ohna, Larget, Liu, Suchard and Huelsenbeck2012), 4 Markov chains were run simultaneously for 10 million generations, with sampling every 1000 generations and a burn-in of 25% to ensure the independence of samples. Stationarity was considered to be reached when the average standard deviation of split frequencies was less than 0.01. Bayesian posterior probabilities (BPP) were estimated on a 50% majority rule consensus tree of the remaining trees. Bootstrap support values were calculated to assess nodal support. We performed a BI phylogenetic analysis based on all codon positions of 13 protein-coding genes (10937 aligned bp) from 81 taxa to establish deep relationships within Orthoptera, which was also used for a divergence time estimate analysis.
Molecular dating requires calibration data, such as a known substitution rates, dated fossil records or palaeogeographical events, or date estimates from previous studies (Magallon, Reference Magallon2004; Sanderson et al., Reference Sanderson, Thorne, Wikstrom and Bremer2004; Drummond et al., Reference Drummond, Ho, Phillips and Rambaut2006; Ho, Reference Ho2007; Ho et al., Reference Ho, Saarma, Barnett, Haile and Shapiro2008), which have different strengths and drawbacks (Heads, Reference Heads2005; Kodandaramaiah, Reference Kodandaramaiah2011). To estimate the time and rates of divergence across major Orthopteran lineages using the split time of 48.7 Mya between Ceracris and Phlaeoba as a calibration point, two fossil calibration constraints were used in the divergence time estimate analysis (Song et al., Reference Song, Amédégnato, Cigliano, Desutter-Grandcolas, Heads, Huang, Otte and Whiting2015). We performed a divergence time estimate analysis using BEAST v.1.8 (Drummond et al., Reference Drummond, Suchard, Xie and Rambaut2012). For this analysis, we used the 81 taxa dataset based on all codon positions of 13 protein-coding genes of complete mitochondrial genome data using the partitioning scheme and the models of nucleotide evolution recommended by PartitionFinder. We created an xml file in BEAUti (Drummond et al., Reference Drummond, Suchard, Xie and Rambaut2012), running a random starting tree, fossil priors, monophyly constraints and parameters for molecular clock models. We used the Yule process as a prior tree. The BI phylogram generated from MrBayes 3.2 was transformed into an ultrametric chronogram using non-parametric rate smoothing (NPRS) in Tree-Edit (Rambaut & Charleston, Reference Rambaut and Charleston2001). This rescaled chronogram was utilized as a random starting tree for BEAST. To assess convergence across independent runs, we conducted two separate analyses, each for 10 million generations, sampling every 1000 generations. A maximum clade credibility tree was summarized in TreeAnnotator (Rambaut & Drummond, Reference Rambaut and Drummond2002–2013b) and visualized in FigTree 1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/) (Rambaut, Reference Rambaut2007).
Results
Mitochondrial genome general features
The complete mitochondrial DNA sequence of C. fasciata was 15569 bp in size (table 2, fig. 1). Nucleotide BLAST (Blastn) of the entire C. fasciata genome against GenBank returned sequence identities with closely related species of 89% (C. versicolor), 89% (C. kiangsu), 87% (Phlaeoba tenebrosa), 87% (Phlaeoba infumata) and 87% (P. albonema) (online table S2). The mitochondrial genome was deposited in the GenBank database under the accession number KP872953. The mitochondrial genome shared the same 37 typical metazoan genes and an A + T-rich region (Flook et al., Reference Flook, Rowell and Gellissen1995b), and they had identical gene arrangement with most species in Caelifera. There were overlapping genes in the mitochondrial genome as in other metazoan mitochondrial genomes (Boore & Brown, Reference Boore and Brown2000). In C. fasciata, the ATG was the most used start codon (eight genes), followed by ATT (one), ATC (two), ACC (one) and ATA (one). The start codon of the cytochrome oxidase subunit I (COX1) gene has been extensively discussed in several arthropod species, including insects. The start codon of the COX1 gene in C. fasciata was ACC. The protein-coding genes take TAA (eight) and TAG (three) as termination codons, except for the NAD5 gene and COX1 (table 2). The two genes had the incomplete termination codon T, which was also found in Locusta migratoria and Gryllotalpa orientalis. The incomplete termination codon was commonly found in metazoan mitochondrial genomes, and a reasonable interpretation was that mRNA polyadenylation makes a complete TAA stop codon. The overlaps occurred 11 times and involved a total of 56 bp. These overlaps existed between ATP8 and ATP6, NAD2 and trnW, COX1 and trnY, COX1 and trnD, NAD3 and trnA, trnG and CO3 on the majority strand, NAD4L and NAD4, NAD4 and trnH, NAD1 and trnL on the minority strand, and between some adjacent genes oriented on opposite strands.
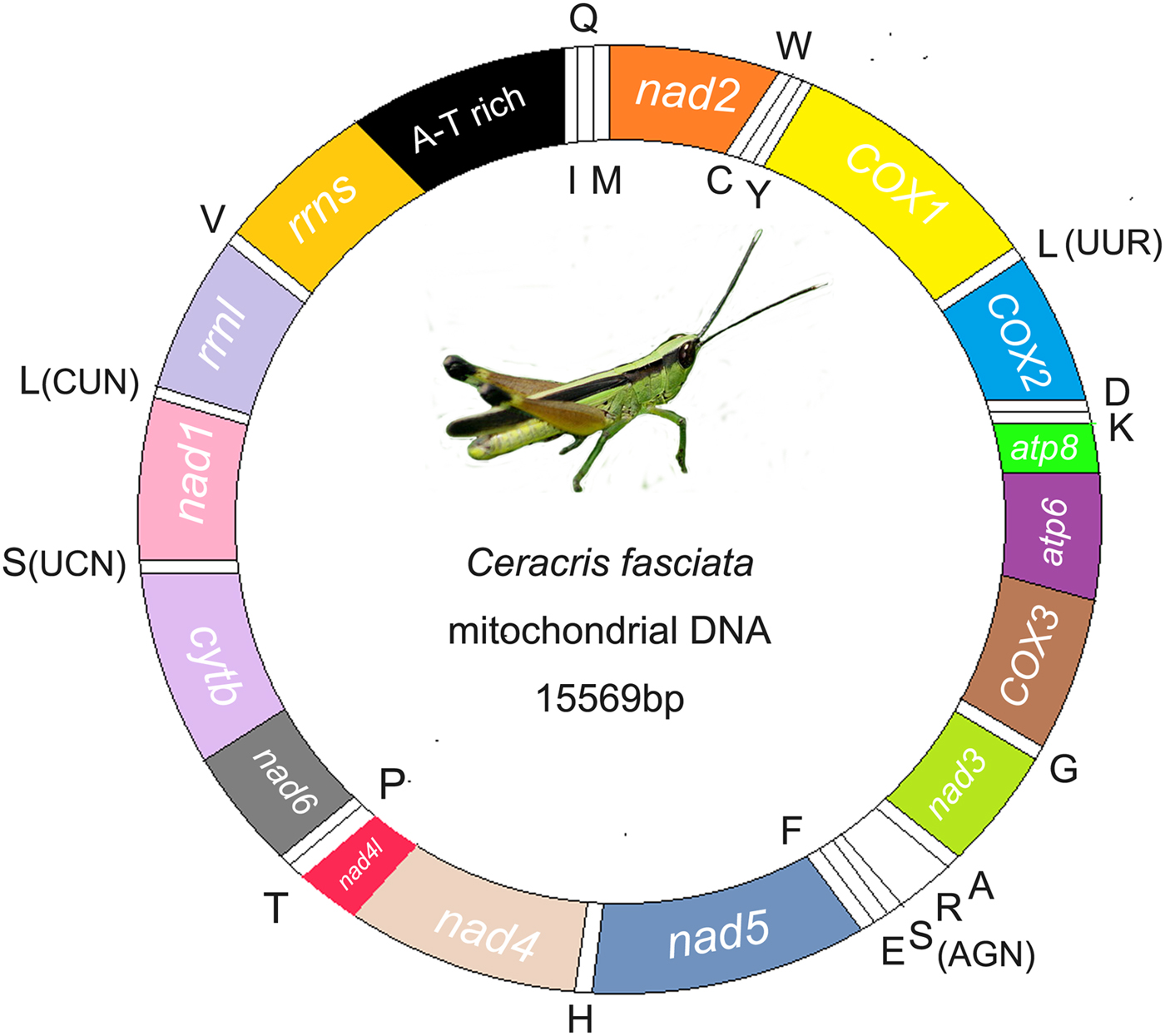
Fig. 1. Graphical representation of the mitochondrial genome of C. fasciata.
Table 2. Annotation of the mitochondrial genome of C. fasciata.
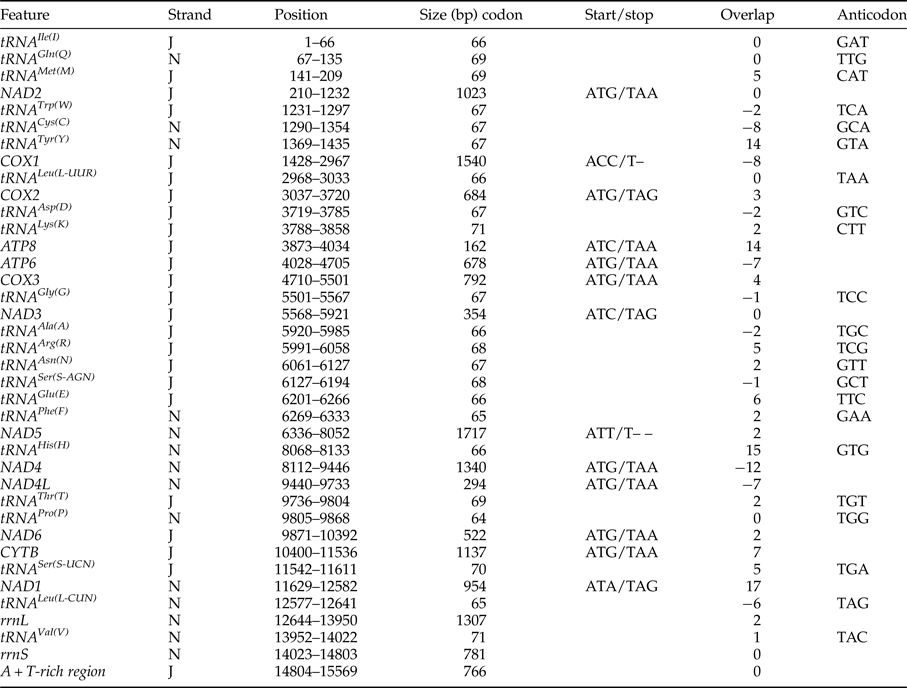
J and N refer to the majority and minority strand, respectively. Position numbers refer to positions on the majority strand.
The nucleotide compositions of the entire mitochondrial DNA sequence for C. fasciata was significantly biased towards A and T. The A + T content was 74.70% (A = 42.63, T = 32.07, C = 14.71, G = 10.57%) in C. fasciata. However, the majority strands of C. fasciata (AT skew = 0.1413, GC skew = −0.1638) favoured A and C. In mammals, the underlying mechanism for this bias of strand-specific nucleotide composition was the deamination of C and A in the H strand during replication (Reyes et al., Reference Reyes, Gissi, Pesole and Saccone1998).
The complete set of 22 tRNA genes typical of metazoan mitochondrial genomes was present in the mitochondrial genome: two for serine and leucine, and one for the other amino acids. All tRNA genes were determined by tRNA-Scan, except for tRNAS (AGN), through the comparison of sequences with previously published Orthopteran mitochondrial genomes. An unusual secondary structure was formed, and it lacked a stable stem-loop structure in the DHU arm. Therefore, the other 21 tRNA genes can be folded into the typical cloverleaf structure (e.g., fig. 2), and their anticodons were identical to those of all the other available Orthopteran mitochondrial genomes. Compared with the published Ensiferan mitochondrial gene order CO2-trnK-trnD-ATP8, there was a rearrangement of trnD and trnK in C. fasciata mitochondrial genomes as well as in those of all the other Caeliferans determined so far. This was consistent with the previous finding that in Orthoptera, such rearrangement occurred only within the Caelifera (Flook et al., Reference Flook, Rowell and Gellissen1995a, Reference Flook, Rowell and Gellissenb). The two genes encoded the large and small ribosomal RNA subunits (rrnL and rrnS), which were located between trnL (CUN) and trnV and between trnV and the A + T-rich region, respectively. The length of rrnL was 1307 bp in C. fasciata, with an A + T content of 77%, and the rrnS was 781 bp in mitochondrial genomes, with an A + T content of 74%.
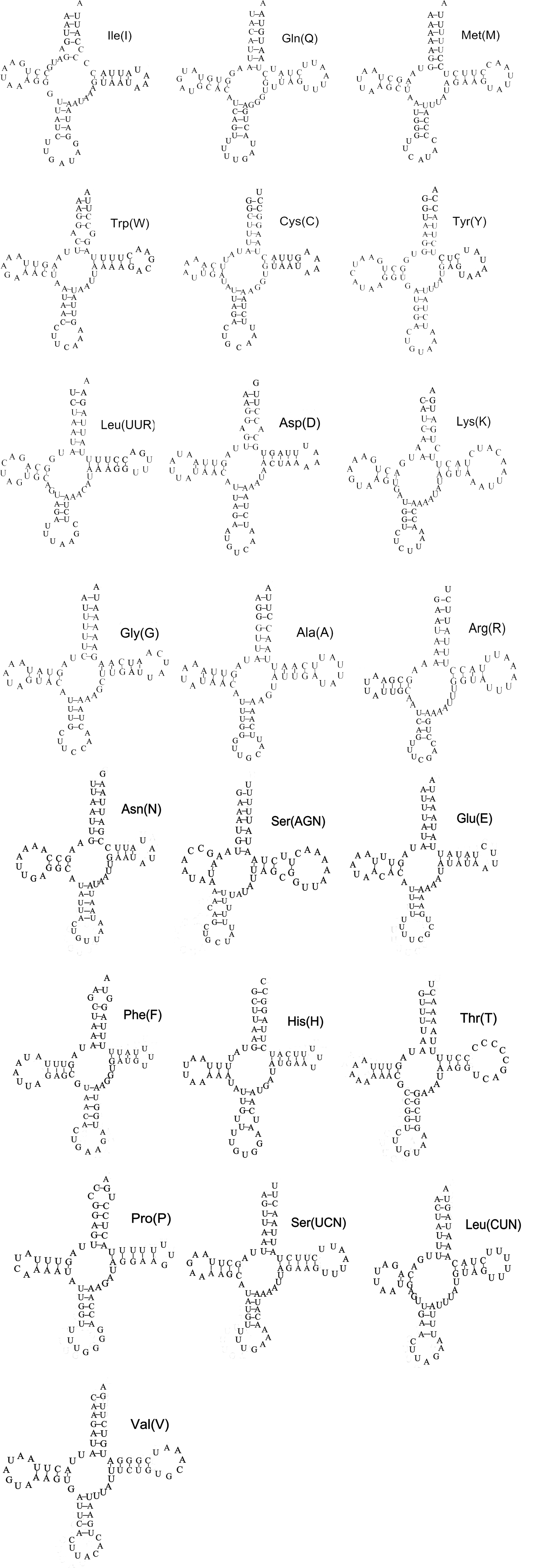
Fig. 2. Inferred secondary structure of 22 tRNAs of C. fasciata. The tRNAs are labeled with the abbreviations of their corresponding amino acids.
All 13 C. fasciata protein-coding genes started with a typical ATN codon, except COX1: one (NAD5) with ATT, two (NAD3, ATP8) with ATC, one (NAD1) with ATA and the other eight (NAD4, NAD4L, CYTB, NAD6, NAD2, COX3, COX2) with ATG (table 2). Previous studies reported no typical ATN initiation codon for COX1 in mitochondrial genomes of many species. As a result, many other anomalous initiation codons, such as ATTA (Van Raay & Crease, Reference Van Raay and Crease1994) and ATTTAA (Piton, Reference Piton1940), were postulated. In C. fasciata, however, the putative start codon for COX1 was ACC, which was located 8 bp upstream of the adjacent trnY. Annotation of a new mitochondrial genome is commonly performed by comparison with closely related mitochondrial genomes that have already been determined. Therefore, the exact start codon for COX1 can finally be determined by protein sequencing.
The 13 protein-coding genes all terminate with the conventional stop codons TAG, TAA or T (table 2). In keeping with other arthropods, overlapping protein-coding genes appeared in the C. fasciata genomes; a 7-bp overlap occurred between ATP8 and ATP6 and between NAD4L and NAD4. In this case, hairpin structures that form at the 3′ end of the upstream protein's mRNA rather than secondary structures of tRNA genes may act as a signal for the cleavage of the polycistronic primary transcript (Fenn et al., Reference Fenn, Cameron and Whiting2007).
The A + T-rich region, which is usually the largest noncoding part of the metazoan mitochondrial DNA molecule, evolves relatively quickly because of few selective constraints. For Orthopterans, a high mutation rate in this region results in significant size variation ranging from 70 bp in the katydid Ruspolia dubia (Zhou et al., Reference Zhou, Huang and Shi2007) to 1512 bp in the grasshopper Chorthippus parallelus (Zhang et al., Reference Zhang, Szymura and Hewitt1995). This variation was predominantly because of both length variations within tandem repeats and differences in their copy numbers. The A + T-rich region contained control elements for the replication and transcription of animal mitochondrial genomes (Boore, Reference Boore1999). Of these elements, the stem-loop secondary structure was potentially involved in the initiation of a second-strand replication (Zhang et al., Reference Zhang, Szymura and Hewitt1995). The possible stem-loop structure located immediately upstream of the origin of the minority strand has been detected in L. Migratoria (Saito et al., Reference Saito, Tmura and Aotsuka2005).
The A + T-rich region in the mitochondrial genome of C. fasciata has a length of 766 bp and was located at the junction rrn S-trnI (fig. 1). There were 78 noncoding nucleotides present in this mitochondrial genome. This region had the highest A + T content, with an A + T content of 85.37%, as well as AT skewness (0.1131) and GC skewness (−0.1607). Conserved elements in the A + T-rich regions were analysed by a comparison with those of Schistocerca gregaria (Zhang et al., Reference Zhang, Szymura and Hewitt1995; Zhang & Hewitt, Reference Zhang and Hewitt1997). We compared the A + T-rich regions of three grasshoppers with each other and found that the percentage of sequence similarity between C. fasciata and S. gregaria was 69.2%, the percentage between C. fasciata and C. versicolor was 78.83%, and the percentage between S. gregaria and C. versicolor was 68.71%. The values for sequence similarity were also high when C. fasciata was compared with other grasshoppers.
Phylogenetic relationships
We performed a phylogenetic analysis with protein sequences of 13 mitochondrial protein-coding genes from 70 in-group Caelifera species and 11 out-group Ensifera species within Orthoptera. ML and BI analyses that used the 13 protein-coding sequences generate tree topologies in the Caelifera suborder (e.g., figs 3 and 4), and our initial analysis using two ribosomal RNA genes sequences led to quite different and confused tree topologies compared with the tree based on all codon positions of protein-coding genes (not shown in figs 3 and 4). Therefore, the analysis that used two ribosomal RNA genes sequences was not suitable in the context of our taxon sampling. The protein-coding sequences are known to be conservative; therefore, it would be more powerful to build a phylogenetic tree this way. Our results may provide evidence for resolving the phylogenetic relationships among the subfamilies and families of Orthoptera. The sister taxon relationship (BPP = 1.00) between Caelifera and Ensifera supported the monophyly of Orthoptera, but not the monophyly of the subfamilies within Caelifera, although the families within Caelifera and the suborders within Ensifera can be recovered as monophyletic. This result was consistent with traditional morphological taxonomy and previous studies (Flook & Rowell, Reference Flook and Rowell1997; Jost & Shaw, Reference Jost and Shaw2006).

Fig. 3. BI phylogenetic analysis based on all codon positions of 13 protein-coding gene. The tree was rooted by Gryllidae, Gryllotalpidae, Myrmecophilidae and Tettigoniidae, Prophalangopsidae, Anostostomatidae, Rhaphidophoridae. Numbers refer to Bayesian posterior probabilities (BPP; near nodes).
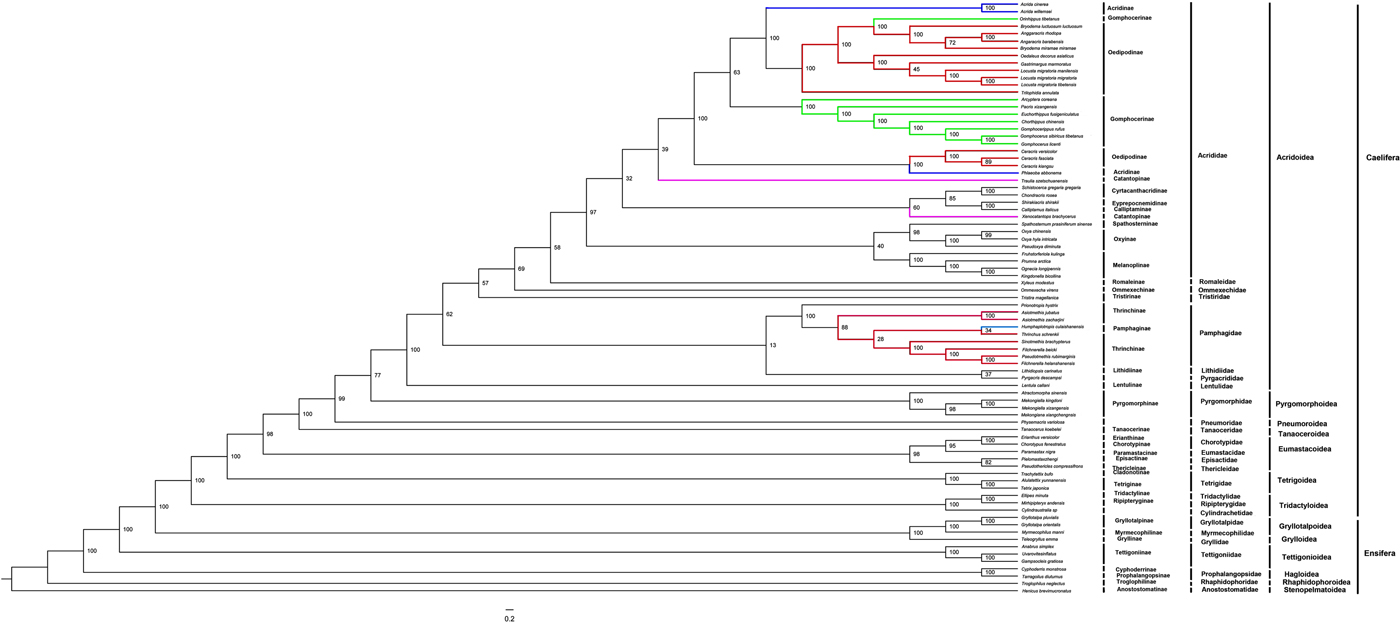
Fig. 4. ML phylogenetic analysis was based on all codon positions of 13 protein-coding genes. The tree was rooted by Gryllidae, Gryllotalpidae, Myrmecophilidae and Tettigoniidae, Prophalangopsidae, Anostostomatidae, Rhaphidophoridae.
The BI phylogenetic analysis, which was based on all codon positions of 13 protein-coding genes, revealed the clades of Caelifera species. Acrididae + Romaleidae forms a group, Ommexechidae is their sister group, and Tristiridae is the external branch for these three families. Lentulidae +Lithidiidae forms a group, and both families are the sister groups of the above four families, while Pamphagidae is their external branch. In addition, Pyrgacrididae is the sister group of the families mentioned above, and Pneumoridae +Pyrgomorphidae are the external branch for all these families. Tanaoceridae is grouped with these families, and then ((Chorotypidae + Eumastacidae) + (Thericleidae + Episactidae)) is the sister group for all the previous families, while the external branch is Tetrigidae. Finally, Ripipterygidae + Tridactylidae and Cylindrachetidae are clustered with all these families for a branch, and the clades of Ensifera have Gryllotaipidae +Myrmecophilidae and Gryllidae as sister groups, with an external branch composed of Tettigoniida, Prophalangopsidae, Rhaphidophoridae and Anostostomatidae.
According to the ML phylogenetic analysis, which was based on all codon positions of 13 protein-coding genes, the result was similar to the BI tree; the classification for Ensifera was completely the same, except Lithidiidae + Pyrgacrididae was identified as a sister group instead of Lentulidae + Lithidiidae, and Pneumoridae was the sister to Pyrgomorphidae and other species groups but not Pyrgomorphidae + Pneumoridae, which formed a sister group in the BI tree. Compared with the failure to recover the monophyletic Orthoptera in previous mitochondrial DNA-based phylogenetic analyses (Kim et al., Reference Kim, Cha, Yoon, Hwang, Lee, Sohn and Jin2005; Carapelli et al., Reference Carapelli, Liò, Nardi, van der Wath and Frati2007), our study showed that analyses with only two species representing the Orthoptera may lead to false phylogenetic inferences.
In total, in the phylogenetic analyses, the species from the families within the suborder Caelifera formed a monophyletic group, as is the case for the Ensifera. However, our results do not support the monophyly of Oedipodinae in Caelifera. Orinhippus tibetanus of Gomphocerinae was always clustered with the species of Oedipodinae in all our phylogeny results, and the BPP were perfect; therefore, we proposed that this species should be classified as a member of Oedipodinae. We found that P. albonema of Acridinae was sorted into a clade with Ceracris species in all our phylogenetic trees; that is, P. albonema was the sister to ((C. fasciata + C. kiangsu) + C. versicolor), and according to field experience, we found that Phlaeoba always lives with Ceracris in the same ecological areas. Therefore, we advise that Phlaeoba should be classified as a member of Oedipodinae. Our initial analysis that used all codon positions of protein-coding genes led to quite similar tree topologies.
In addition, we also found that Humphaplotropis culaishanensis of Pamphaginae was always clustered with species of Thrinchinae in all our results; therefore, we propose that H. culaishanensis should be classified as a member of Thrinchinae. For C. fasciata, we found that it was always clustered with C. kiangsu, and these two species were sisters of C. versicolor. Therefore, the genetic relationship between C. fasciata and C. kiangsu was closer than that between C. fasciata and C. versicolor.
Divergence time estimation
The estimated ages were also shown on the calibrated ML tree (fig. 5). The oldest estimated divergence time of Ensifera in this study was 146.16 Mya, around the late Jurassic or early Cretaceous. We estimated that katydids (Grylloidea) likely diverged from other groups in the early Cretaceous. According to our divergence time estimation analyses, we concluded that the ancestral Acrididae probably originated in the early Paleogene, and it is likely that the major diversification events happened at the middle Paleogene, well into the next geologic time. We estimated that crickets (Tettigoniidae) diverged from other groups probably in the early Cretaceous. Acrididae and Romaleinae, the youngest two clades we observed, were estimated to have diverged 56.7 Mya between the middle and higher Paleogene. The Pyrgacrididae and Ommexechidae were clustered in a group that was a sister to the Acrididae. The divergence time between Acrididae and Romaleinae group and Pyrgacrididae and Ommexechidae group is 58.79. C. versicolor was sister to the C. kiangsu and C. fasciata group. First, C. versicolor diverged from the sister group (C. kiangsu + C. fasciata) around 44.81 Mya, then C. kiangsu and C. fasciata separated from each other 43.04 Mya.
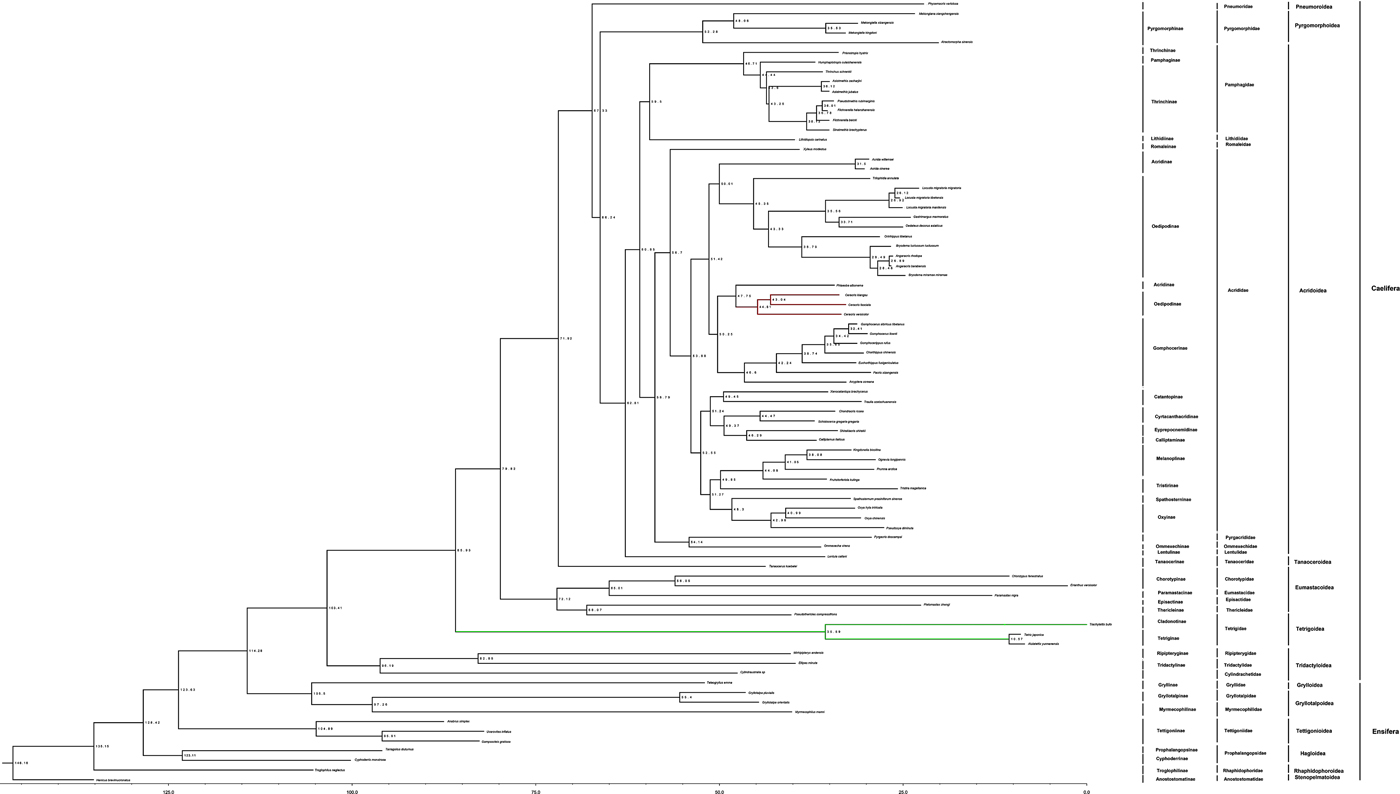
Fig. 5. A divergence time-estimate analysis of Orthoptera based on mtgenome data and calibration points using BEAST. The numbers next to the nodes are the estimated node ages. Terminals are coded to show family-level relationships. Family names used in this figure follow the currently recognized 26 families according to the Orthoptera Species File (Cigliano et al., Reference Cigliano, Braun, Eades and Otte2017).
Discussion
Mitochondrial genome general features
At the family level, the length of the C. fasciata mitochondrial genome (15,569 bp) was marginally smaller than that of C. kiangsu (15,665 bp), although it falls within the range (15,971–18,133 bp) of other known Orthopteran mitochondrial genomes in GenBank. The gene order and orientation were the same as in previously sequenced Acrididae. The nucleotide BLAST (Blastn) results of the entire mitochondrial genome against closely related species revealed that C. fasciata has a high similarity with Acrididae species (87% in Gonista bicolor–89% in C. versicolor) (online table S2). The conserved regions lie in 22 tRNAs and 13 PCGs, while the A + T-rich region varies in this species. These remarkable characteristics have been reported in other Orthopteran species (Kim et al., Reference Kim, Cha, Yoon, Hwang, Lee, Sohn and Jin2005) and could be used as potential markers for identification at the genus and species levels using recent molecular techniques. The high A + T-biased nucleotide composition was similar to that of previously sequenced Orthopteran species (75.23% in C. versicolor; 74.89% in C. kiangsu; 76.28% in G. bicolor; and 74.08% in P. infumata). The positive AT skew (0.1413) observed here, indicating the presence of more As than Ts, was similar to that seen in many Orthopteran species, including C. versicolor (0.1304), C. kiangsu (0.1562), P. infumata (0.1527) and G. bicolor (0.1369) and was slightly lower than that of other sequenced mitochondrial genomes in Pyrgomorphoidea, including Atractomorpha sinensis (0.1601), Mekongiella kingdoni (0.1608) and Mekongiana xiangchengensis (0.1463). We also observed GC skew (−0.1638) here, which was similar to that in other animals. The A + T-rich region also revealed the higher A + T content, a common feature in Caelifera, which may imply that the higher mutation rate of this region was related to higher A + T content and under lower selection pressure (Yang et al., Reference Yang, Du, Wang, Cao and Yu2011).
Phylogenetic relationships
This work also represents the most comprehensive phylogenetic analysis of Orthoptera to date (figs 4 and 5) and presents an excellent opportunity to test previous hypotheses about the phylogenetic relationships among the major lineages within the order. Next, we will comment on higher-level relationships, which we can confidently resolve using current data. Our results were consistent with the list provided by Song et al. (Reference Song, Amédégnato, Cigliano, Desutter-Grandcolas, Heads, Huang, Otte and Whiting2015), which proposed a phylogeny-based natural classification scheme for Orthoptera (table 3). We now have a very clear understanding of the phylogeny of Caelifera and the relationships among superfamilies and families. Previous molecular phylogenetic hypotheses for Caelifera (Flook & Rowell, Reference Flook and Rowell1997; Flook et al., Reference Flook, Klee and Rowell1999; Leavitt et al., Reference Leavitt, Hiatt, Whiting and Song2013; Zhang et al., Reference Zhang, Huang, Lin, Wang and Zheng2013a, Reference Zhang, Zeng, Huang and Zhengb) consistently recovered the basal position of Tridactyloidea sensu Dirsh (Reference Dirsh1975), followed by Tetrigoidea sensu Dirsh (Reference Dirsh1975), and our study also confirmed this hypothesis. Although earlier studies that focused on the fossil evidence proposed a sister relationship between Tridactyloidea and Tetrigoidea (Ragge, Reference Ragge1955; Sharov, Reference Sharov1968), other morphologists have suggested that Tetrigoidea was more closely related to other Caeliferan superfamilies (Kevan, Reference Kevan and Parker1982; Dirsh, Reference Dirsh1975; Vickery, Reference Vickery, Gangwere, Muralirangan and Muralirangan1997), and our study concurred with the latter. Tridactyloidea was the earliest branch within Caelifera and currently consists of three families: Tridactylidae (pygmy mole crickets), the Neotropical relative Ripipterygidae, and the subterranean Cylindrachetidae (sandgropers). Although Vickery (Reference Vickery1997) considered that Cylindrachetidae formed its own superfamily, Cylindrachaetoidea, Flook & Rowell (Reference Flook and Rowell1997) included it within Tridactyloidea, and our results supported this position. Acridoidea was the largest superfamily within Acridomorpha and currently includes 11 recognized families and more than 8000 described species, which were defined by the morphology of the male phallic complex and a missing basioccipital slit, among other characteristics (Roberts, Reference Roberts1941; Chopard, Reference Chopard and Grasse1949; Dirsh, Reference Dirsh1973; Amedegnato, Reference Amedegnato1974; Eades, Reference Eades2000). Many species in this group can be recognized as typical and familiar grasshoppers. Within Acridoidea, we found that Pyrgacrididae, which was endemic to Reunion Island in the Indian Ocean (Hugel, Reference Hugel2005), was the earliest diverging lineage and represented a transitional form between Pyrgomorphoidea and Acridoidea (Eades, Reference Eades2000). Because we focused on the Caelifera, we did not analyse Ensiferan sequences phylogenetically in detail, although we tried to include all the eligible Orthopteroid orders in our samples. The primary Ensiferan radiation appears to precede that of the Caelifera, and this observation was consistent with an analysis of a mitochondrial tRNA gene rearrangement of trnD and trnK in Orthoptera (Reyes et al., Reference Reyes, Gissi, Pesole and Saccone1998) and our results. The present study represents a major result for Orthopteran systematics and provides a framework for a phylogeny-based natural classification system for Orthoptera (table 1). Our study also could help to clarify the phylogenetic position for Rhaphidophoroidea, which includes a single cosmopolitan family, Rhaphidophoridae (camel crickets). Gorochov (Reference Gorochov and Field2001) considered Rhaphidophoridae to be a member of Stenopelmatoidea, but our study showed that it is divergent from Stenopelmatoidea, and our results were consistent with those of Song et al. (Reference Song, Amédégnato, Cigliano, Desutter-Grandcolas, Heads, Huang, Otte and Whiting2015).
Table 3. A phylogeny-based natural classification scheme proposed in this study.

Although this study did not illustrate long-standing problems, it identified several areas of research that require further investigation. The first area is the relationship among basal Ensifera. Our taxon sampling in these groups was insufficient, and these taxa are also among the least studied groups within Orthoptera. Similarly, more sampling for Gryllidea is required to formally test the validity of Gryllotalpoidea and the relationships among the families and to check the definition of monophyletic families within Grylloidea. The second area is to entirely re-evaluate the classification of Tettigoniidae. The katydid classification is clearly in a state of flux, and a major revision of the classification will be required once a definitive phylogeny of Tettigoniidae becomes available. Thus, in this study, we recognized a single family, Tettigoniidae, which has always been found to be monophyletic, and this result was consistent with that of Song et al. (Reference Song, Amédégnato, Cigliano, Desutter-Grandcolas, Heads, Huang, Otte and Whiting2015). However, the number of species chosen in this study was less than that used in their study, so more specimens are needed to re-examine the phylogenic relationships within Tettigoniidae. The third area is the phylogeny of Tetrigoidea, which is one of the more ancient lineages and the second most speciose superfamily within Caelifera. Tetrigoidea had a universal distribution and showed exceptional diversity in pronotum morphology. However, the phylogeny of Tetrigoidea is completely unknown at the moment.
The phylogeny of Ensifera has been contentious over the years, and numerous hypotheses have been proposed based on different character systems (Judd, Reference Judd1947; Blackith & Blackith, Reference Blackith and Blackith1968; Ragge, Reference Ragge1977; Gorochov, Reference Gorochov1995a; Gwynne, Reference Gwynne1995; Desutter-Grandcolas, Reference Desutter-Grandcolas2003; Eades et al., Reference Eades, Otte, Cigliano and Braun2014). Within the infraorder Gryllidea, we have determined Gryllidae to be a sister to a clade that consists of Gryllotalpidae and Myrmecopholidae. The close relationship between Gryllidae and Gryllotalpidae has always been supported by previous studies (Ander, Reference Ander1939; Zeuner, Reference Zeuner1939; Roberts, Reference Roberts1941; Judd, Reference Judd1947; Vickery, Reference Vickery1977; Song et al., Reference Song, Amédégnato, Cigliano, Desutter-Grandcolas, Heads, Huang, Otte and Whiting2015). Our phylogeny showed that Gryllidae is a stable sister to Myrmecophilidae + Gryllotalpidae. Larger taxon and character sampling of the total evidence analysis will enable us to overcome this issue in the future.
Divergence time estimation
Orthoptera has evolved over 300 Myr (Sharov, Reference Sharov1968; Gorochov, Reference Gorochov1995a; Storozhenko, Reference Storozhenko, Gangwere, Muralirangan and Muralirangan1997; Grimaldi & Engel, Reference Grimaldi and Engel2005), and its current diversity has been shaped by dynamic shifts of diversification rates at different geological times across different lineages within the order. Certain branches are diverse and show an international distribution, while other branches are represented by only a few species and have a limited geographical range (Kevan, Reference Kevan and Parker1982). In addition, different evolutionary branches show characteristic morphological, ecological and behavioural traits that appear to be conserved in each lineage (Kevan, Reference Kevan and Parker1982).
Using the divergence time estimates, we discussed the evolution of the three most diverse and cosmopolitan Orthopteran clades, crickets (Grylloidea), katydids (Tettigoniidae), and grasshoppers (Acrididae), to highlight the complexity of Orthopteran evolution.
Katydids represent the most successful lineage within Orthoptera with respect to species diversity (Eades, Reference Eades2000). We estimated that they likely diverged from other groups in the early Cretaceous. They are also very diverse in terms of ecological traits because their diets range from herbivory to carnivory and they can be active both during the day and at night (Gwynne, Reference Gwynne2001). Similar to crickets, katydids communicate acoustically, with their songs ranging from audible sound to ultrasound (Greenfield, Reference Greenfield, Gangwere, Muralirangan and Muralirangan1997; Gwynne, Reference Gwynne2001; Montealegre-Z, Reference Montealegre-Z2009), with very rare loss of acoustic communication, and sexual selection has likely played an important role in their diversification. However, definitive fossils of Tettigoniidae are known only from the Cenozoic (Piton, Reference Piton1940; Sharov, Reference Sharov1968; Gorochov, Reference Gorochov1995b; Storozhenko, Reference Storozhenko, Gangwere, Muralirangan and Muralirangan1997).
Grasshoppers represent the most recently diverged lineage within Orthoptera. Fossil acridids are frustratingly scarce, and most of the known fossils are from the mid- to late Cenozoic (Sharov, Reference Sharov1968; Storozhenko, Reference Storozhenko, Gangwere, Muralirangan and Muralirangan1997; Grimaldi & Engel, Reference Grimaldi and Engel2005). According to our divergence time estimation analyses, the time that Acrididae separate from other families is 56.7 Mya, this result is later than that in Song et al. (Reference Song, Amédégnato, Cigliano, Desutter-Grandcolas, Heads, Huang, Otte and Whiting2015). We concluded that the ancestral Acrididae probably originated in the early Paleogene, and it is likely that the major diversification events happened at the middle Paleogene, well into the next geologic time. C. versicolor was sister to the C. kiangsu and C. fasciata group. First of all, C. versicolor diverged from the sister group (C. kiangsu + C. fasciata) around 44.81 Mya, then C. kiangsu and C. fasciata separated from each other 43.04 Mya. Phlaeoba separate from Ceracris is 47.75 Mya, this result is almost in keeping with that result from Song et al. (Reference Song, Amédégnato, Cigliano, Desutter-Grandcolas, Heads, Huang, Otte and Whiting2015). The most speciose acridid subfamily is Gomphocerinae (Eades et al., Reference Eades, Otte, Cigliano and Braun2014), which has mouthparts adapted for graminivory. Thus, it is conceivable that the diversification of grasses, and in particular the spread of open grasslands (Dixon et al., Reference Dixon, Faber-Langendoen, Josse, Morrison and Loucks2014), could have played an important role in the diversification of grasshoppers (Dixon et al., Reference Dixon, Faber-Langendoen, Josse, Morrison and Loucks2014).
Crickets represent one of the most ancient lineages within Orthoptera. They form the third most diverse clade, with more than 4800 known species (Eades, Reference Eades2000). We estimated that they diverged from other groups probably in the early Cretaceous.
Modern crickets are mostly nocturnal, brown or black in colour, and omnivorous scavengers (Alexander, Reference Alexander1968), and these ecological characters could have originated in the early Mesozoic. What sets the crickets apart from other ancient insects of similar ecological habits is their ability to communicate acoustically (Greenfield, Reference Greenfield, Gangwere, Muralirangan and Muralirangan1997), although the loss of acoustic communication is frequent in crickets compared with tettigoniids. Cricket songs are mainly used in the context of intraspecific sexual behaviour (Otte, Reference Otte1992; Greenfield, Reference Greenfield, Gangwere, Muralirangan and Muralirangan1997), which suggests that acoustic crickets are under sexual selection. Many closely related species can be diagnosed by their male songs. Sexually selected characteristics tend to evolve rapidly (Lande, Reference Lande1981; West-Eberhard, Reference West-Eberhard1983) and, in fact, the effect of sexual selection on male calling songs has been postulated as a likely reason for the extremely rapid speciation in a Hawaiian genus, Laupala (Mendelson & Shaw, Reference Mendelson and Shaw2005). At the same time, these songs can attract predators and parasitoids, which can provide strong selective pressure for the crickets to rapidly evolve silence (Pascoal et al., Reference Pascoal, Cezard, Eik-Nes, Gharbi, Majewska, Payne, Ritchie, Zuk and Bailey2014) or to modify their signals into less conspicuous ones, such as the ultrasound signals used by Eneopterinae (Robillard et al., Reference Robillard, Grandcolas and Desutter-Grandcolas2007). Therefore, both sexual selection and natural selection for songs may have played important roles in cricket diversification over the past 200 Myr, which may have contributed to the current diversity (Song et al., Reference Song, Amédégnato, Cigliano, Desutter-Grandcolas, Heads, Huang, Otte and Whiting2015).
Orthoptera is the hemimetabolous insect assemblage and still contains many species that are underrepresented by complete mitochondrial sequences. Since our taxon sampling was relatively weak compared with that of other groups included in this study, additional sampling may be necessary to test those hypotheses. With advances in sequencing technologies (Shendure & Ji, Reference Shendure and Ji2008), a new approach to phylogenetic studies utilizing the whole genome and transcriptome data, also known as phylogenomics, is becoming increasingly popular (Faircloth et al., Reference Faircloth, McCormac, Crawford, Harvey, Brumfield and Glenn2012; Lemmon et al., Reference Lemmon, Emme and Lemmon2012). For Orthoptera, though the order is known to have the largest nuclear genome size among insects (Hanrahan & Johnston, Reference Hanrahan and Johnston2011), ranging from 1.52 to 16.56 Gb (Gregory, Reference Gregory2014), the L. migratoria genome have been fully annotated and its official gene set is freely available online (http://159.226.67.243/). A phylogenomic analysis using universal single-copy genes of locusts and other insects has been performed (Wang et al., Reference Wang, Fang, Yang, Jiang, Jiang, Zhao, Li, Cui, Wei, Ma, Wang, He, Luo, Wang, Guo, Guo, Zhang, Yang, Hao, Chen, Ma, Yu, Xiong, Zhu, Fan, Han, Wang, Chen, Wang, Yang, Zhao, Feng, Chen, Lian, Li, Huang, Yao, Lv, Zhang, Li, Zhu and Kang2014). Currently, the gene function for unknown sequences can be determined by homology searches using known protein sequences from sequenced insects (even distantly related specie). Therefore, the absence of a reference Orthopteran genome do not makes gene annotation for any shotgun sequencing or RNA-seq difficult. Alternatively, with the progress of modern science and technology, transcriptome-based analysis represents a cheap and efficient way, and single-copy conserved genes are feasible to be assembled by transcriptome data (Misof et al., Reference Misof, Liu, Meusemann, Peters, Donath, Mayer, Frandsen, Ware, Flouri, Beutel, Niehuis, Petersen, Izquierdo-Carrasco, Wappler, Rust, Aberer, Aspöck, Aspöck, Bartel, Blanke, Berger, Böhm, Buckley, Calcott, Chen, Friedrich, Fukui, Fujita, Greve, Grobe, Gu, Huang, Jermiin, Kawahara, Krogmann, Kubiak, Lanfear, Letsch, Li, Li, Li, Lu, Machida, Mashimo, Kapli, McKenna, Meng, Nakagaki, Navarrete-Heredia, Ott, Ou, Pass, Podsiadlowski, Pohl, von Reumont, Schütte, Sekiya, Shimizu, Slipinski, Stamatakis, Song, Su, Szucsich, Tan, Tan, Tang, Tang, Timelthaler, Tomizuka, Trautwein, Tong, Uchifune, Walzl, Wiegmann, Wilbrandt, Wipfler, Wong, Wu, Wu, Xie, Yang, Yang, Yeates, Yoshizawa, Zhang, Zhang, Zhang, Zhang, Zhao, Zhou, Zhou, Ziesmann, Zou, Li, Xu, Zhang, Yang, Wang, Wang, Kjer and Zhou2014). So it is not a puzzle to phylogenomic studies for Orthoptera. Then we will attempt to use transcriptome data analyse Orthoptera phylogeny next. We wish for a much more thorough understanding of the evolution of this fascinating group of insects. This work has added to current knowledge on the hemipteran phylogeny inferred from mitochondrial genomes. Future research that integrates more taxon samplings, more mitochondrial genome sequences, and data from other molecular markers will provide greater insights into the evolution of Orthoptera.
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0007485317000761.
Acknowledgements
Special thanks to Si-Yu Dong, Wan-Wei Dong and Ran Li for technical assistance and advice on the experiments. This work was supported by grants from the National Natural Sciences Foundation of China (Grant numbers 30970339 and 31572246) to Professor Guo-Fang Jiang.










