



Introduction
Although only 0.3% of Continental Antarctica is ice-free, Antarctica is home to many organisms including bacteria, unicellular eukaryotes, fungi, lichen, cryptogamic plants and invertebrates that are scattered across the continent and subsist in isolated, remote, island-like habitats (Convey et al. Reference Convey, Chown, Clarke, Barnes, Bokhorst, Cummings, Ducklow, Frati, Green, Gordon, Griffiths, Howard-Williams, Huiskes, Laybourn-Parry, Lyons, McMinn, Morley, Peck, Quesada, Robinson, Schiaparelli and Wall2014), for example, in soils, lakes and cryoconite holes. Availability of biodiversity information from these Antarctic areas is required for three major reasons. First, such data facilitate the investigation of glacial constraints and effects on current biodiversity (Convey et al. Reference Convey, Stevens, Hodgson, Smellie, Hillenbrand, Barnes, Clarke, Pugh, Linse and Cary2009). Second, it allows investigation of the effects of environmental change on Antarctic ecosystems (Nielsen & Wall Reference Nielsen and Wall2013), and finally, conservation management becomes possible, also in light of increasing threats from non-indigenous invasive species (Chown et al. Reference Chown, Huiskes, Gremmen, Lee, Terauds, Crosbie, Frenot, Hughes, Imura, Kiefer, Lebouvier, Raymond, Tsujimoto, Ware, van de Vijver and Bergstrom2012a, Reference Chown, Lee, Hughes, Barnes, Barrett, Bergstrom, Convey, Cowan, Crosbie, Dyer, Frenot, Grant, Herr, Kennicutt, Lamers, Murray, Possingham, Reid, Riddle, Ryan, Sanson, Shaw, Sparrow, Summerhayes, Terauds and Wall2012b, Reference Chown, Hodgins, Griffin, Oakeshott, Byrne and Hoffmann2015b). However, knowledge of terrestrial Antarctic biodiversity is still limited because the vast majority of Antarctica’s ice-free areas remain un- or under-studied (McGaughran et al. Reference McGaughran, Stevens, Hogg and Carapelli2011, Convey et al. Reference Convey, Chown, Clarke, Barnes, Bokhorst, Cummings, Ducklow, Frati, Green, Gordon, Griffiths, Howard-Williams, Huiskes, Laybourn-Parry, Lyons, McMinn, Morley, Peck, Quesada, Robinson, Schiaparelli and Wall2014).
Biodiversity research of ice-free habitats in Antarctica is complicated. First, logistic difficulties exacerbated by the harsh environmental conditions may limit biological research to the proximity of research stations, when more extensive field work is required (Convey Reference Convey2010). Second, traditional biodiversity assessments of many multicellular eukaryotes include manual sorting and morphological identification, which are time consuming and require specific taxonomic expertise, especially for the cryptic and inconspicuous terrestrial life of Antarctica (e.g. Velasco-Castrillón et al. Reference Velasco-Castrillón, Gibson and Stevens2014a). Molecular methods are better suited to the study of Antarctic biota (Rogers Reference Rogers2007), but may lack resolution when sequence information is not considered (e.g. the analysis of terminal restriction fragment length polymorphisms or similar techniques; Magalhaes et al. Reference Magalhaes, Stevens, Cary, Ball, Storey, Wall, Tuerk and Ruprecht2012, Makhalanyane et al. Reference Makhalanyane, Valverde, Birkeland, Cary, Tuffin and Cowan2013, Dreesens et al. Reference Dreesens, Lee and Cary2014) or may be labour intensive (e.g. Sanger sequencing; Lawley et al. Reference Lawley, Ripley, Bridge and Convey2004, Fell et al. Reference Fell, Scorzetti, Connell and Craig2006, Velasco-Castrillón & Stevens Reference Velasco-Castrillón and Stevens2014, Velasco-Castrillón et al. Reference Velasco-Castrillón, Page, Gibson and Stevens2014b, Reference Velasco-Castrillón, Schultz, Colombo, Gibson, Davies, Austin and Stevens2014c).
Readily applied in many other parts of the world (reviewed in Bik et al. Reference Bik, Porazinska, Creer, Caporaso, Knight and Thomas2012a, Bohmann et al. Reference Bohmann, Evans, Gilbert, Carvalho, Creer, Knapp, Yu and de Bruyn2014), metabarcoding approaches (sensu Taberlet et al. Reference Taberlet, Coissac, Pompanon, Brochmann and Willerslev2012a) present an opportunity to rapidly generate baseline biodiversity information for a variety of terrestrial Antarctic habitats (Fig. 1; Chown et al. Reference Chown, Hodgins, Griffin, Oakeshott, Byrne and Hoffmann2015b). Metabarcoding approaches use the genetic material from bulk environmental samples such as soil, permafrost, water, ice, snow or other substrates (Bohmann et al. Reference Bohmann, Evans, Gilbert, Carvalho, Creer, Knapp, Yu and de Bruyn2014). Then DNA from multiple organisms contained in such samples are identified for taxonomic analyses either with traditional Sanger sequencing or more recently using high-throughput sequencing (HTS; Chown et al. Reference Chown, Hodgins, Griffin, Oakeshott, Byrne and Hoffmann2015b, Cowan et al. Reference Cowan, Ramond, Makhalanyane and de Maayer2015, Czechowski et al. Reference Czechowski, Clarke, Breen, Cooper and Stevens2016). In a global context, HTS-supported metabarcoding approaches have been applied to monitoring invasive species and surveying biodiversity over large spatial scales (Drummond et al. Reference Drummond, Newcomb, Buckley, Xie, Dopheide, Potter, Heled, Ross, Tooman, Grosser, Park, Demetras, Stevens, Russell, Anderson, Carter and Nelson2015 and reviewed in Bohmann et al. Reference Bohmann, Evans, Gilbert, Carvalho, Creer, Knapp, Yu and de Bruyn2014). In Antarctica, metabarcoding studies, initially based on Sanger sequencing, have enabled the identification of cryptic organisms and communities such as fungi, yeast and invertebrates (Lawley et al. Reference Lawley, Ripley, Bridge and Convey2004, Fell et al. Reference Fell, Scorzetti, Connell and Craig2006). These techniques have also been applied to viruses (López-Bueno et al. Reference López-Bueno, Tamames, Velázquez, Moya, Quesada and Alcamí2009), bacteria in hypolithic communities, soil and air (Makhalanyane et al. Reference Makhalanyane, Valverde, Birkeland, Cary, Tuffin and Cowan2013, Bottos et al. Reference Bottos, Scarrow, Archer, McDonald and Cary2014a, Reference Bottos, Woo, Zawar-Reza, Pointing and Cary2014b), as well as fungal and unicellular eukaryotes of soils (Dreesens et al. Reference Dreesens, Lee and Cary2014, Niederberger et al. Reference Niederberger, Sohm, Gunderson, Parker, Tirindelli, Capone, Carpenter and Cary2015). Additionally, the methodological pitfalls of these techniques when applied in Antarctica have become better understood (Lee et al. Reference Lee, Herbold, Polson, Wommack, Williamson, McDonald and Cary2012b, Czechowski et al. Reference Czechowski, Clarke, Breen, Cooper and Stevens2016), including amplification and sequencing biases, coupled with sparse reference data. Collectively, HTS metabarcoding, despite not being without flaws, provides a promising method to rapidly gather biodiversity information from Antarctic habitats, with the ability to generate large amounts of biodiversity data from a wide range of taxa with simple sample collection, uniform laboratory workflows and comparable data structures.
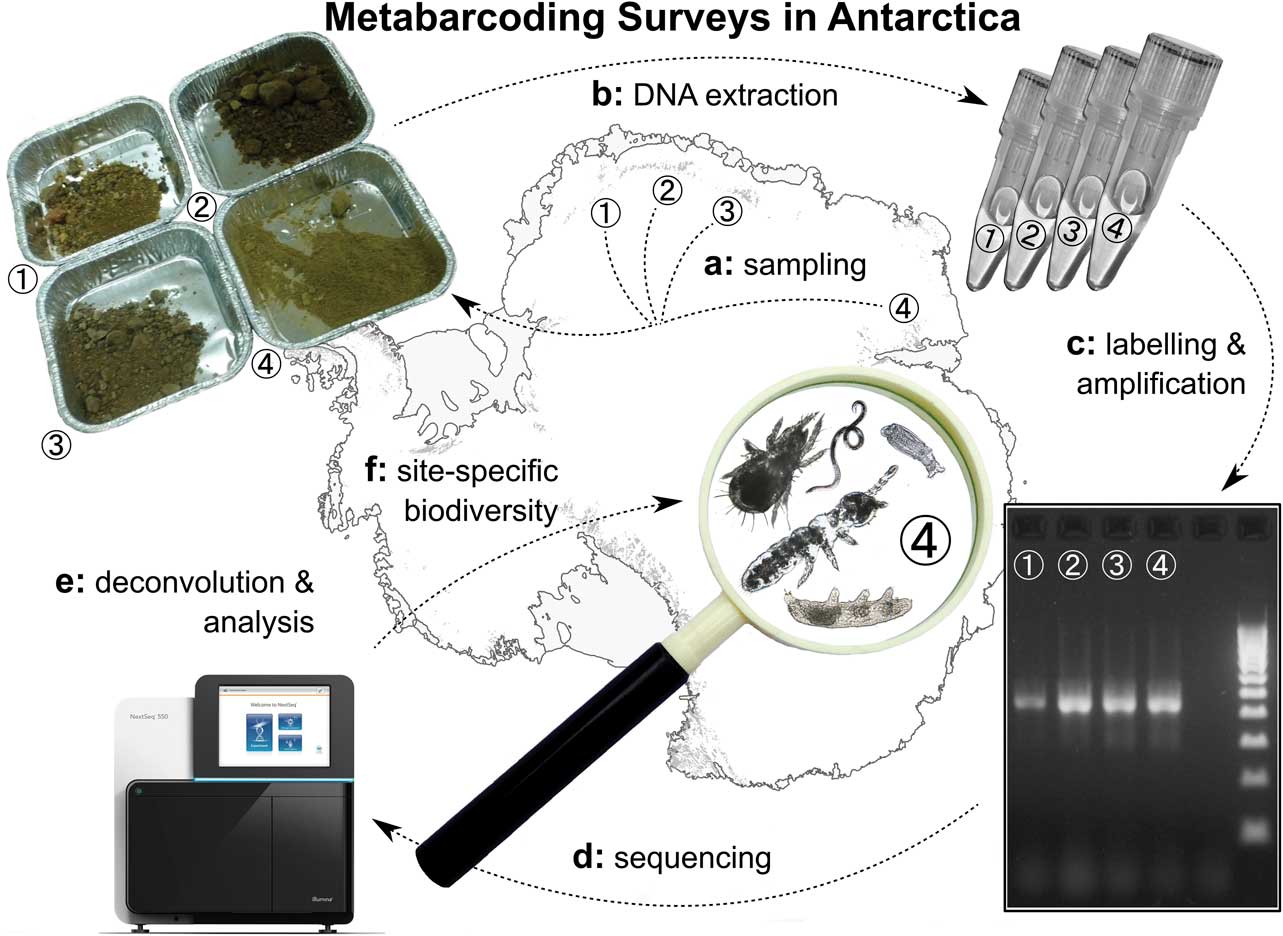
Fig. 1 Workflow for metabarcoding analyses, which can be applied to soil, snow, ice, cryconite holes, lake sediments or nearshore marine environments. a. Samples are collected. b. The genetic material is extracted in bulk from individual samples. c. DNA contained in extracts is amplified with genetic markers and sequencing adapters, multiplex identifier (MID) tags are added. d. The library is processed on a high-throughput sequencing device. e. After data deconvolution according to sample, reference information assigns individual sequences or sequence clusters with taxonomic information. f. Distributional information becomes available. Picture of sequencing device provided courtesy of Illumina (San Diego, CA, USA). Base layers courtesy of the Scientific Committee on Antarctic Research Antarctic Digital Database.
Here, we provide a technical introduction to HTS metabarcoding with an Antarctic focus and highlight the potential of such approaches for Antarctic biodiversity research beyond their current applications. This synthesis serves as a starting point for the development of Antarctic HTS metabarcoding surveys. We hope to encourage fellow researchers to participate in the joint effort of understanding Antarctica’s biodiversity on a continental scale (Kennicutt et al. Reference Kennicutt, Chown, Cassano, Liggett, Massom, Peck, Rintoul, Storey, Vaughan, Wilson and Sutherland2014).
Technical considerations
Metabarcoding projects are influenced by biases inherent to several methodological aspects. These include: i) extraction of representative DNA from a mixed template sample and the intra- and extracellular DNA contained in such a sample, ii) platform-specific sequencing technologies including inherent sequence error patterns, iii) the appropriate choice of markers, iv) methods for generation and v) amplification of sequencing libraries. Finally, informed approaches to HTS data processing and analysis are necessary to achieve research goals.
Sample selection
As shown in a variety of global studies (reviewed in Bohmann et al. Reference Bohmann, Evans, Gilbert, Carvalho, Creer, Knapp, Yu and de Bruyn2014), it is possible to extract DNA suitable for metabarcoding analyses from a variety of substrates, which offers a unique opportunity to study different environments in Antarctica (Fig. 1a). DNA can be extracted from organisms contained in surface soil (Czechowski et al. Reference Czechowski, Clarke, Breen, Cooper and Stevens2016), permafrost (Bellemain et al. Reference Bellemain, Davey, Kauserud, Epp, Boessenkool, Coissac, Geml, Edwards, Willerslev, Gussarova, Taberlet and Brochmann2013), snow (Dalén et al. Reference Dalén, Götherström, Meijer and Shapiro2007), ice (Willerslev et al. Reference Willerslev, Hansen and Poinar2004), freshwater benthos of lakes (Hajibabaei et al. Reference Hajibabaei, Spall, Shokralla and van Konynenburg2012) or nearshore marine sediments (Powell et al. Reference Powell, Bowman, Snape and Stark2003). Furthermore, extracts of pre-sorted samples can be analysed (Drummond et al. Reference Drummond, Newcomb, Buckley, Xie, Dopheide, Potter, Heled, Ross, Tooman, Grosser, Park, Demetras, Stevens, Russell, Anderson, Carter and Nelson2015), such as from museum collections. When limited starting material is available, preservatives such as ethanol can be used as a DNA source (Shokralla et al. Reference Shokralla, Singer and Hajibabaei2010). DNA can also be extracted from faeces (Jarman et al. Reference Jarman, McInnes, Faux, Polanowski, Marthick, Deagle, Southwell and Emmerson2013), for example the seal and penguin colonies in coastal regions of Antarctica and sub-Antarctic islands. The variety of potential sample types, coupled with cost-effective sequence data generation, could address problems related to surveying large spatial or temporal scales.
Extraction of environmental samples
Failure to extract representative DNA from a sample (Fig. 1b), so-called ‘extraction bias’ (Pedersen et al. Reference Pedersen, Overballe-Petersen, Ermini, Sarkissian, Haile, Hellstrom, Spens, Thomsen, Bohmann, Cappellini, Schnell, Wales, Caroe, Campos, Schmidt, Gilbert, Hansen, Orlando and Willerslev2014), is a major concern for metabarcoding approaches. Such biases occur when extraction methods inconsistently lyse cells of different organisms, and are further biased by the presence of dead organisms’ DNA in substrates (Pedersen et al. Reference Pedersen, Overballe-Petersen, Ermini, Sarkissian, Haile, Hellstrom, Spens, Thomsen, Bohmann, Cappellini, Schnell, Wales, Caroe, Campos, Schmidt, Gilbert, Hansen, Orlando and Willerslev2014). Some authors explicitly distinguish an intracellular DNA component from an extracellular DNA component in bulk extracts, and present methods to quantify both fractions in a given sample (Ascher et al. Reference Ascher, Ceccherini, Pantani, Agnelli, Borgogni, Guerri, Nannipieri and Pietramellara2009). Yet, applying such approaches across large numbers of samples may be cost-prohibitive. Alleviating DNA extraction biases can be achieved through combining different extraction methods, and include blending samples prior to extraction and/or using a large amount of starting material (Delmont et al. Reference Delmont, Robe, Cecillon, Clark, Constancias, Simonet, Hirsch and Vogel2011, Reference Delmont, Simonet and Vogel2013, Taberlet et al. Reference Taberlet, Prud’Homme, Campione, Roy, Miquel, Shehzad, Gielly, Rioux, Choler, Clement, Melodelima, Pompanon and Coissac2012b). Yet, different extraction methods or batch-wise application of one extraction method may introduce variable levels of non-template contamination (Salter et al. Reference Salter, Cox, Turek, Calus, Cookson, Moffatt, Turner, Parkhill, Loman and Walker2014). Therefore, randomized drawing of sample batches is recommended (Salter et al. Reference Salter, Cox, Turek, Calus, Cookson, Moffatt, Turner, Parkhill, Loman and Walker2014). Extraction biases and contamination can be discovered by inclusion of negative and positive controls. Negative controls facilitate the detection of contamination. Positive controls of known taxonomic composition are helpful in detecting compositional deviations between the sequence data and sample source (Salter et al. Reference Salter, Cox, Turek, Calus, Cookson, Moffatt, Turner, Parkhill, Loman and Walker2014, Czechowski et al. Reference Czechowski, Clarke, Breen, Cooper and Stevens2016). Consequently, both positive and negative controls help to optimize the DNA extraction process and are helpful in streamlining processing parameters in steps following extraction.
High-throughput sequencing platforms
The recent advance of HTS-supported metabarcoding, metagenetics and metagenomics (Bik et al. Reference Bik, Porazinska, Creer, Caporaso, Knight and Thomas2012a, Taberlet et al. Reference Taberlet, Coissac, Pompanon, Brochmann and Willerslev2012a, Bohmann et al. Reference Bohmann, Evans, Gilbert, Carvalho, Creer, Knapp, Yu and de Bruyn2014) can be considered a consequence of continuing development of sequencing platforms by companies such as 454 (Roche, Basel, official platform support was discontinued in 2016), Illumina (San Diego, CA, USA), IonTorrent (Thermo Fisher Scientific, Waltham, MA, USA) and others since 2005 (Glenn Reference Glenn2011, van Dijk et al. Reference Van Dijk, Auger, Jaszczyszyn and Thermes2014). These devices generate substantially larger amounts of sequencing data than chain-termination sequencing (Bohmann et al. Reference Bohmann, Evans, Gilbert, Carvalho, Creer, Knapp, Yu and de Bruyn2014), but in comparison produce shorter reads (i.e.~100–800 base pairs, depending on the technology). Using these platforms in conjunction with metabarcoding (and metagenomic) approaches removes the need to process mixed DNA templates through clone libraries and hence substantially reduces the time to data generation. The platform of choice to conduct metabarcoding biodiversity surveys currently appears to be one of the Illumina platforms, due to the large number of sequences generated which reduces the cost per base and the comparatively low error rate of this sequencing platform (Bokulich et al. Reference Bokulich, Subramanian, Faith, Gevers, Gordon, Knight, Mills and Caporaso2013, Bragg et al. Reference Bragg, Stone, Butler, Hugenholtz and Tyson2013). The discontinued 454 platform, although often comparatively expensive to use, will continue for some time to offer the longest read lengths of all platforms suitable for amplicon sequencing (Van Dijk et al. Reference Van Dijk, Auger, Jaszczyszyn and Thermes2014). Comprehensive reviews of HTS platforms are provided in Glenn (Reference Glenn2011) and van Dijk et al. (Reference Van Dijk, Auger, Jaszczyszyn and Thermes2014). Currently, the most common and cost-effective approach to generate metabarcoding information with HTS is parallel sequencing of PCR-amplified bulk DNA extracts, known as ‘amplicon sequencing’ (Taberlet et al. Reference Taberlet, Coissac, Pompanon, Brochmann and Willerslev2012a, Bohmann et al. Reference Bohmann, Evans, Gilbert, Carvalho, Creer, Knapp, Yu and de Bruyn2014). Important methodological aspects of amplicon sequencing are described below. We also describe how pitfalls of amplicon sequencing can be alleviated and present alternative methods for library generation and sequencing.
Marker choice
Markers for PCR amplification of mixed DNA templates extracted from environmental samples (e.g. soil (Fig. 1c), permafrost, water, ice, snow, etc.) should i) ideally amplify all taxa with similar efficiency despite potential mismatches between primers and the variety of template molecules (Clarke et al. Reference Clarke, Soubrier, Weyrich and Cooper2014a), ii) amplify target regions short enough to allow amplification of degraded DNA, particularly if targeting extracellular DNA (Riaz et al. Reference Riaz, Shehzad, Viari, Pompanon, Taberlet and Coissac2011, Coissac et al. Reference Coissac, Riaz and Puillandre2012, Taberlet et al. Reference Taberlet, Coissac, Pompanon, Brochmann and Willerslev2012a), iii) exhibit the least possible amount of degenerate bases to allow the application of high annealing temperatures, while decreasing the risk of chimeric amplification (Lenz & Becker Reference Lenz and Becker2008, Ahn et al. Reference Ahn, Kim, Song and Weon2012) and iv) target a gene region for which ample reference data are available to allow taxonomic identification of phylotypes (see below).
Finding a primer pair that possesses these desirable, possibly incompatible, qualities is challenging. Two genes that have been widely applied in single-gene and metabarcoding analyses of metazoans, for example, are the nuclear 18S ribosomal DNA (18S rDNA) and mitochondrial cytochrome c oxidase subunit I (COI) genes (Wu et al. Reference Wu, Ayres, Bardgett, Wall and Garey2011, Zhan et al. Reference Zhan, Bailey, Heath and Macisaac2014). These markers are favoured due to their long history of application, resulting in comparatively abundant reference data in sequence repositories such as GenBank, BOLD and SILVA (Pruesse et al. Reference Pruesse, Quast, Knittel, Fuchs, Ludwig, Peplies and Gloeckner2007, Ratnasingham & Hebert Reference Ratnasingham and Hebert2007, Benson et al. Reference Benson, Karsch-Mizrachi, Lipman, Ostell and Sayers2011) (Fig. 1e & f). However, 18S rDNA data may underestimate biodiversity due to low taxonomic resolution, and many COI markers show inherent taxonomic bias due to insufficiently conserved primer binding sites across broad taxonomic groups (Tang et al. Reference Tang, Leasi, Obertegger, Kieneke, Barraclough and Fontaneto2012, Deagle et al. Reference Deagle, Jarman, Coissac, Pompanon and Taberlet2014). Similar advantages and disadvantages are found analogously in other marker regions applied in metabarcoding studies, for example when targeting fungi using the ITS region or photosynthetic cryptogams via the matK and chloroplast genes (CBOL Plant Working Group 2009, Orgiazzi et al. Reference Orgiazzi, Bianciotto, Bonfante, Daghino, Ghignone, Lazzari, Lumini, Mello, Napoli, Perotto, Vizzini, Bagella, Murat and Girlanda2013, Drummond et al. Reference Drummond, Newcomb, Buckley, Xie, Dopheide, Potter, Heled, Ross, Tooman, Grosser, Park, Demetras, Stevens, Russell, Anderson, Carter and Nelson2015).
Library generation
Preparing DNA for HTS requires the addition of platform-specific sequencing adapters, and often (particularly for metabarcoding) sample-specific sequence tags (or ‘multiplex identifier’ (MID) tags) are also required to enable deconvolution of sequence data (Fig. 1c & e). Initially, DNA pools were furnished with MID tags during PCR (Saiki et al. Reference Saiki, Gelfand, Stoffel, Scharf, Higuchi, Horn, Mullis and Erlich1988) via extended primer sequences or ligation of unmodified primers preceding sequence adapter ligation (Binladen et al. Reference Binladen, Gilbert, Bollback, Panitz, Bendixen, Nielsen and Willerslev2007, Meyer et al. Reference Meyer, Stenzel and Hofreiter2008). More recently, library generation via long primer sequences carrying both sequence adaptor and MID tags (fusion primers) has become common (Bik et al. Reference Bik, Porazinska, Creer, Caporaso, Knight and Thomas2012a). The application of fusion primers is practical in that it only requires a single PCR, but may be costly for large numbers of samples and difficult for primer lengths above ~50 base pairs due to poor PCR performance. In those cases, more labour intensive ligation protocols may be a better choice (Stiller et al. Reference Stiller, Knapp, Stenzel, Hofreiter and Meyer2009, Kircher et al. Reference Kircher, Sawyer and Meyer2012, O’Neill et al. Reference O’Neill, Schwartz, Bullock, Williams, Shaffer, Aguilar-Miguel, Parra-Olea and Weisrock2013). Also of concern is the informed choice of MID tags. Owing to possible flaws in the underlying algorithms, these tags may not meet the intended expectations of robustness towards sequencing errors (Faircloth & Glenn Reference Faircloth and Glenn2012). Only MID tags that have been explicitly tested for correct Hamming distances (Hamming Reference Hamming1950) are recommended, and this will later enable correct deconvolution and error correction (Faircloth & Glenn Reference Faircloth and Glenn2012).
Amplification
Concordance between the taxonomic composition of a mixed DNA template retrieved from environmental bulk samples and the amplified library requires careful calibration of PCR conditions, for example, length optimization of denaturation, annealing and extension steps as well as the correct temperatures for the primer annealing phase (Fig. 1c). Possible pitfalls include i) introduction of substitutions and insertion/deletions through polymerase activity (Cline et al. Reference Cline, Braman and Hogrefe1996), ii) formation of chimeric molecules in late amplification stages (Kanagawa Reference Kanagawa2003), iii) amplification bias when using degenerate primers in combination with high annealing temperatures (Cline et al. Reference Cline, Braman and Hogrefe1996, Kanagawa Reference Kanagawa2003), and iv) failure to detect rare variants when little replication is applied (Ficetola et al. Reference Ficetola, Pansu, Bonin, Coissac, Giguet-Covex, De Barba, Gielly, Lopes, Boyer, Pompanon, Raye and Taberlet2015). Such pitfalls collectively threaten the credibility of the resulting sequence data (Czechowski et al. Reference Czechowski, Clarke, Breen, Cooper and Stevens2016). They may i) alter the similarity of phylotypes to reference sequences, ii) result in artificial phylotypes that match several reference sequences, iii) artificially enrich phylotypes whose library molecules matched the PCR primers well or iv) result in false-negative concealment of phylotypes. Retrieval of higher quality data can be achieved by i) application of proofreading polymerases (Taberlet et al. Reference Taberlet, Coissac, Pompanon, Brochmann and Willerslev2012a), ii) using few and long PCR cycles (Kanagawa Reference Kanagawa2003, Lenz & Becker Reference Lenz and Becker2008, Ahn et al. Reference Ahn, Kim, Song and Weon2012), iii) careful testing of annealing temperature (Sipos et al. Reference Sipos, Székely, Palatinszky, Révész, Márialigeti and Nikolausz2007) and iv) processing three or more PCR replicates (Gilbert et al. Reference Gilbert, Meyer, Antonopoulos, Balaji, Brown, Desai, Eisen, Evers, Field, Feng, Huson, Jansson, Knight, Knight, Kolker, Konstantindis, Kostka, Kyrpides, Mackelprang, McHardy, Quince, Raes, Sczyrba, Shade and Stevens2010). Analogous to the extraction step, positive and negative controls are important to track contamination during the amplification procedure (Czechowski et al. Reference Czechowski, Clarke, Breen, Cooper and Stevens2016). At the same time, positive controls may be a source of cross-contamination, for example, through unintended PCR-product carry-over (Kwok Reference Kwok1990). Using suitable non-Antarctic control DNA, which can be distinguished from sample DNA in later analysis steps, could reduce the impact of PCR-product carry-over.
Sequence analysis
Most importantly, it needs to be noted that processing HTS data (Fig. 1e & f) is not straightforward and requires a high level of bioinformatics expertise, project-specific software selection and software fine-tuning at every step. To perform a metabarcoding analysis with any given raw dataset, first an analysis workflow needs to be conceptualized. Then, a variety of software algorithms need to be selected with regard to the analysis steps and study goals, keeping in mind available computing hardware, methods of library design, employed sequencing technology, data volume and analysis pitfalls (Coissac et al. Reference Coissac, Riaz and Puillandre2012, Lee et al. Reference Lee, Herbold, Polson, Wommack, Williamson, McDonald and Cary2012b). Subsequently, testing programs individually and in order of application using small datasets is advisable. Here, it may be necessary to generate custom (or at least modify existing) scripts, through which data input and output of algorithms is handled and connected.
It is possible that the resulting metabarcoding analysis workflow is initiated by marker selection (Riaz et al. Reference Riaz, Shehzad, Viari, Pompanon, Taberlet and Coissac2011), and once sequence data has been generated, several raw data processing steps will follow before the statistical analysis can be attempted (Bik et al. Reference Bik, Porazinska, Creer, Caporaso, Knight and Thomas2012a, Bohmann et al. Reference Bohmann, Evans, Gilbert, Carvalho, Creer, Knapp, Yu and de Bruyn2014). Raw data preparation typically includes quality filtering, removal of sequence adapters, data deconvolution and chimera removal. The clean raw data are then typically clustered, assigned with taxonomy, and subsequently, the resulting data are checked for their suitability for the intended statistical analysis.
Although raw data preparation can be achieved with a variety of programs (see Table I for examples), software environments dedicated to metabarcoding analysis such as QIIME, MOTHUR and MG-RAST (Schloss et al. Reference Schloss, Westcott, Ryabin, Hall, Hartmann, Hollister, Lesniewski, Oakley, Parks, Robinson, Sahl, Stres, Thallinger, van Horn and Weber2009, Caporaso et al. Reference Caporaso, Kuczynski and Stombaugh2010, Wilke et al. Reference Wilke, Bischof, Gerlach, Glass, Harrison, Keegan, Paczian, Trimble, Bagchi, Gram, Chaterji and Meyer2016) offer functionality incorporating whole analysis workflows starting from raw data cleaning, phylotype clustering and basic statistical analyses. These metabarcoding/metagenomic software environments themselves usually take advantage of multiple algorithms dedicated to particular sub-routines of analysis workflows. For example, chimera detection may be achieved with UPARSE (Edgar Reference Edgar2013) in QIIME. Taxonomic assignments may be retrieved with BLAST (Altschul et al. Reference Altschul, Gish, Miller, Myers and Lipman1990) or other algorithms such as the RDP classifier, or UCLUST (Edgar Reference Edgar2010, Lan et al. Reference Lan, Wang, Cole and Rosen2012). In general, sub-algorithms employed by software suites need to be carefully considered before attempting data preparation and analysis steps. An overview of raw data preparation software is provided by Zhou & Rokas (Reference Zhou and Rokas2014).
Table I Selection of analysis software for metabarcoding data of environmental DNA. Possible tasks related to handling of metabarcoding data provided in columns. Multi-purpose tools in top section are suitable for various sequence analysis tasks. Software environments in the middle section are specialized for metabarcoding analysis. Programs and packages in the bottom section are focussed on ecological analysis and/or visualizing results. X=functionality of a given tool for a given task is provided, (X)=functionality of a given tool for a given task is provided to some extent, -=functionality is not provided.

If dedicated metabarcoding/metagenomic analysis environments do not offer desired functionalities for analysis and visualization, some analyses can be achieved through other available software and possibly linked in via ‘glue code’ written in programming languages such as R (R Development Team 2016), BASH or PYTHON (Van Rossum & Drake Reference Van Rossum and Drake1995). EXPLICET (Robertson et al. Reference Robertson, Harris, Wagner, Granger, Browne, Tatem, Feazel, Park, Pace and Frank2013), for example, offers basic visualization and statistical analysis functionally coupled with a graphical user interface, suitable for novice users. More powerful, but command-driven, the R environment offers several packages for the statistical analysis and visualization of metabarcoding data with packages such as PHYLOSEQ or VEGAN (McMurdie & Holmes Reference McMurdie and Holmes2013, Oksanen et al. Reference Oksanen, Blanchet, Kindt, Legendre, Minchin, O’Hara, Simpson, Solymos, Stevens and Wagner2015, R Development Team 2016) and many others. An in-depth review of metabarcoding and metagenomic sequence analysis software is provided by Lindgreen et al. (Reference Lindgreen, Adair and Gardner2016).
Biodiversity surveys often seek to quantify α-diversity (species richness) and β-diversity (change in community composition; Whittaker Reference Whittaker1960). Three pitfalls should be carefully considered when designing metabarcoding analysis workflows aiming at α- and β-diversity comparisons between a given set of samples, and while relating species occurrences to, for example, their environment. All analyses require a) sufficient sequencing depth, b) α- and β-diversity comparisons require appropriate abundance correction of libraries with different read depths and c) distance-based ordination techniques can only be applied appropriately when using the correct distance measure. Regarding a) above, it is important to realize that sequencing depth of HTS libraries is crucial for the reliable estimation of biodiversity measures in the resulting data (Smith & Peay Reference Smith and Peay2014); while in order to retrieve reliable biodiversity estimates from Antarctic habitats, a high sequencing effort may be necessary to retrieve credible statistical results (Czechowski et al. Reference Czechowski, Clarke, Breen, Cooper and Stevens2016). Regarding b) above, the analysis of differentially abundant phylotypes in metabarcoding data, by comparisons of proportions or rarefied counts, although applied widely, is inappropriate and may yield misleading results (McMurdie & Holmes Reference McMurdie and Holmes2014). Instead of applying such rarefaction methods, other algorithms should be employed to enable comparison between libraries with coverage differences. For instance, R packages DESeq and edgeR offer alternative ways to correct phylotype abundance (Robinson et al. Reference Robinson, McCarthy and Smyth2009, Anders & Huber Reference Anders and Huber2010). Regarding c) above, scarcity of biological data is known to impair ecological statistical analysis in Antarctica due to the low spatial overlap of individual phylotypes (Magalhaes et al. Reference Magalhaes, Stevens, Cary, Ball, Storey, Wall, Tuerk and Ruprecht2012, Czechowski et al. Reference Czechowski, Clarke, Breen, Cooper and Stevens2016). Consequently, metabarcoding data from many Antarctic habitats is likely to be difficult to analyse with commonly used distance-based ordination methods including multidimensional scaling (Wish & Carroll Reference Wish and Carroll1982), constrained analysis of principal components (CAP) and redundancy analysis (RDA) (Legendre & Andersson Reference Legendre and Andersson1999). Hence, when employing distance-based ordination approaches, sample comparison may only be possible with metrics established to be suitable, for example the Hellinger distance (Gagné & Proulx Reference Gagné and Proulx2009). Alternatively, several different distance metrics should be compared (Blanchet et al. Reference Blanchet, Legendre, Bergeron and He2014). Model-based ordination methods may circumvent drawbacks of distance-based ordinations, and should be used where possible (Ellis et al. Reference Ellis, Smith and Pitcher2012, Wang et al. Reference Wang, Naumann, Wright and Warton2012, Hui et al. Reference Hui, Taskinen, Pledger, Foster and Warton2015).
Recent improvements of high-throughput sequencing metabarcoding
Retrieving biodiversity information from hundreds of samples over large spatial or temporal scales requires cost-efficient processing. Tagging individual samples with fusion primers for amplicon sequencing is simple, but increases the cost of HTS metabarcoding studies for large-scale approaches. Presumably for this reason, numbers of parallel processed samples in several recent global and Antarctic metabarcoding studies range from seven to twelve samples (Bik et al. Reference Bik, Sung, De Ley, Baldwin, Sharma, Rocha-Olivares and Thomas2012b, Roesch et al. Reference Roesch, Fulthorpe, Pereira, Pereira, Lemos, Barbosa, Suleiman, Gerber, Pereira, Loss and da Costa2012, Dreesens et al. Reference Dreesens, Lee and Cary2014, Niederberger et al. Reference Niederberger, Sohm, Gunderson, Parker, Tirindelli, Capone, Carpenter and Cary2015). Reducing primer-associated costs is possible through modular combination of multiple sequence tags per sample, thus reducing the amount of unique oligonucleotides required for a project. Examples of such modular workflows include using two PCRs to double-tag amplicons for HTS (Bybee et al. Reference Bybee, Bracken-Grissom, Haynes, Hermansen, Byers, Clement, Udall, Wilcox and Crandall2011, de Cárcer et al. Reference De Cárcer, Denman, McSweeney and Morrison2011). Similarly, double-tagging can generate amplicons with minimal work, handling and cost in a single PCR (Clarke et al. Reference Clarke, Czechowski, Soubrier, Stevens and Cooper2014b).
The PCR biases during library preparation can be alleviated through the application of hybridization approaches. In hybridization approaches, libraries are generated by annealing target DNA to biotinylated oligonucleotide probes (Gnirke et al. Reference Gnirke, Melnikov, Maguire, Rogov, LeProust, Brockman, Fennell, Giannoukos, Fisher, Russ, Gabriel, Jaffe, Lander and Nusbaum2009, Faircloth et al. Reference Faircloth, McCormack, Crawford, Harvey, Brumfield and Glenn2012, Lemmon et al. Reference Lemmon, Emme and Lemmon2012). In comparison to PCR, hybridization approaches enable retrieval of multiple conserved regions per reaction, perform well in detecting rare DNA and reduce compositional biases in the resulting data without the need for extensive replication (Taberlet et al. Reference Taberlet, Coissac, Pompanon, Brochmann and Willerslev2012a). For example, Denonfoux et al. (Reference Denonfoux, Parisot, Dugat-Bony, Biderre-Petit, Boucher, Morgavi, Le Paslier, Peyretaillade and Peyret2013) sequenced bacterial DNA derived from environmental samples after enrichment with a hybridization approach, demonstrating the benefits outlined here for mixed template DNA sources.
The lengths of genomic regions that can be targeted with single read lengths of a given HTS platform are variable (see section ‘High-throughput sequencing platforms’), but usually shorter than the 600–1000 base pairs that can be achieved from a single read using Sanger sequencing technology. Therefore, recent research has investigated the options of adopting shorter fragments of regions that have been used widely in Sanger sequencing, for example, the beginning of the COI gene region or the 18S gene (Machida & Knowlton Reference Machida and Knowlton2012, Leray et al. Reference Leray, Yang, Meyer, Mills, Agudelo, Ranwez, Boehm and Machida2013). Other studies have identified new marker regions with short read lengths suitable for HTS technologies, which retain adequate information allowing comparisons with data from the traditional markers (CBOL Plant Working Group 2009, Epp et al. Reference Epp, Boessenkool, Bellemain, Haile, Esposito, Riaz, Erseus, Gusarov, Edwards, Johnsen, Stenoien, Hassel, Kauserud, Yoccoz, Brathen, Willerslev, Taberlet, Coissac and Brochmann2012). Furthermore, identification of custom marker regions is now possible with bioinformatics tools such as ecoPrimers incorporated into OBI tools (Riaz et al. Reference Riaz, Shehzad, Viari, Pompanon, Taberlet and Coissac2011, Boyer et al. 2016) (see Table I). ecoPrimers employs user-curated reference data retrieved from repositories such as GenBank (Benson et al. Reference Benson, Karsch-Mizrachi, Lipman, Ostell and Sayers2011) to identify conserved regions suitable for project-specific primer design for mixed template amplification.
Other methods of streamlining metabarcoding approaches with regard to data yields and cost efficiency have become available. A combination of shotgun sequencing methods and amplicon sequencing, for instance, allow retrieval of full length COI sequences using HTS technology (Liu et al. Reference Liu, Li, Lu, Su, Tang, Zhang, Zhou, Zhou, Yang, Ji, Yu and Zhou2013). Furthermore, the omission of library quantification, and instead pooling libraries by volume (coupled with shearing and re-assembly; Feng et al. Reference Feng, Liu, Chen, Liang and Zhang2015), can reduce time and effort during library construction. Finally, with decreasing sequencing costs, metagenomic studies targeting the entirety of DNA molecules in a sample, including functional genes, without selective amplification or enrichment (Fierer et al. Reference Fierer, Leff, Adams, Nielsen, Bates, Lauber, Owens, Gilbert, Wall and Caporaso2012) may become viable for large sample numbers.
The potential of metabarcoding and metagenomics for Antarctic biology
Elucidating community structures
Community-level interaction is an important feature of Antarctic ecosystems. Such interactions were believed to be minimal, perhaps owing to the fact that they are hard to measure (Hogg et al. Reference Hogg, Cary, Convey, Newsham, O’Donnell, Adams, Aislabie, Frati, Stevens and Wall2006). However, biotic community-level interactions are increasingly implicated in facilitating survival in harsh environments, and may be observable through stratified occurrence of different organisms or the exchange of nutrients between strata within communities (Nakai et al. Reference Nakai, Abe, Baba, Imura, Kagoshima, Kanda, Kohara, Koi, Niki, Yanagihara and Naganuma2012, Pointing & Belnap Reference Pointing and Belnap2012). Community-level organization has been discovered among Antarctic soil crusts, lithobiontic communities, eukaryotes in moss pillars and cyanobacterial mats (Jungblut et al. Reference Jungblut, Vincent and Lovejoy2012, Nakai et al. Reference Nakai, Abe, Baba, Imura, Kagoshima, Kanda, Kohara, Koi, Niki, Yanagihara and Naganuma2012, Makhalanyane et al. Reference Makhalanyane, Valverde, Birkeland, Cary, Tuffin and Cowan2013, Colesie et al. Reference Colesie, Gommeaux, Green and Bueddel2014). Evidence for biotic interactions has also been reported among soil arthropods of sub-Antarctic islands (Caruso et al. Reference Caruso, Trokhymets, Bargagli and Convey2013).
Studies describing the community-level organization of Antarctic terrestrial ecosystems will benefit from metabarcoding and metagenomic approaches. Possible studies could include further analyses of prokaryotic and eukaryotic diversity in substrates such as snow, soil crusts and hypolithons, photobiotic and mycobiotic diversity and biogeography of lichen, or the association between fungi and eukaryotes in moss communities, that are still often studied using Sanger sequencing (Carpenter et al. Reference Carpenter, Lin and Capone2000, Fernández-Mendoza et al. Reference Fernández-Mendoza, Domaschke, García, Jordan, Martín and Printzen2011, Khan et al. Reference Khan, Tuffin, Stafford, Cary, Lacap, Pointing and Cowan2011, Jungblut et al. Reference Jungblut, Vincent and Lovejoy2012, Gokul et al. Reference Gokul, Valverde, Tuffin, Cary and Cowan2013, Altermann et al. Reference Altermann, Leavitt, Goward, Nelsen and Lumbsch2014). The HTS-supported analysis of such communities is becoming more common for eukaryotes and bacteria, e.g. in cyanobacterial mats and hypolithic communities (Lee et al. Reference Lee, Barbier, Bottos, McDonald and Cary2012a, Dreesens et al. Reference Dreesens, Lee and Cary2014, Niederberger et al. Reference Niederberger, Sohm, Gunderson, Parker, Tirindelli, Capone, Carpenter and Cary2015). However, similar approaches could be applied to environments such as air (Bottos et al. Reference Bottos, Woo, Zawar-Reza, Pointing and Cary2014b) or nearshore sediments (Powell et al. Reference Powell, Bowman, Snape and Stark2003). Functional aspects of soil microbial communities have been investigated using metagenomic HTS approaches, providing an in-depth picture of ecosystem services (Fierer et al. Reference Fierer, Leff, Adams, Nielsen, Bates, Lauber, Owens, Gilbert, Wall and Caporaso2012). Similarly, HTS-based metagenomic studies advanced the description of morphologically conserved, rare or small cryptic communities in Antarctica, including their provision of ecosystem services (Goordial et al. Reference Goordial, Davila, Greer, Cannam, DiRuggiero, McKay and Whyte2016).
Supporting conservation of Antarctica
The biodiversity and distribution of the terrestrial Antarctic biota is more heterogeneous than anywhere else in the world (Ettema & Wardle Reference Ettema and Wardle2002, Convey et al. Reference Convey, Chown, Clarke, Barnes, Bokhorst, Cummings, Ducklow, Frati, Green, Gordon, Griffiths, Howard-Williams, Huiskes, Laybourn-Parry, Lyons, McMinn, Morley, Peck, Quesada, Robinson, Schiaparelli and Wall2014, Chown et al. Reference Chown, Clarke, Fraser, Cary, Moon and McGeoch2015a). Large distances between habitats, unique geological and glacial histories, different soil compositions and extreme fluctuations of abiotic conditions amplify this heterogeneity (Bockheim Reference Bockheim1997, Marchant & Head Reference Marchant and Head2007, Bintanja et al. Reference Bintanja, Severijns, Haarsma and Hazeleger2014). Consequently, Antarctic biota exhibit a high degree of endemism and costly adaptation mechanisms to withstand harsh environmental conditions (Convey Reference Convey1997, Convey & Stevens Reference Convey and Stevens2007). Human-mediated environmental changes are anticipated to have profound effects on the spatial extent and structure of Antarctic terrestrial ecosystems (Chown et al. Reference Chown, Huiskes, Gremmen, Lee, Terauds, Crosbie, Frenot, Hughes, Imura, Kiefer, Lebouvier, Raymond, Tsujimoto, Ware, van de Vijver and Bergstrom2012a, Reference Chown, Lee, Hughes, Barnes, Barrett, Bergstrom, Convey, Cowan, Crosbie, Dyer, Frenot, Grant, Herr, Kennicutt, Lamers, Murray, Possingham, Reid, Riddle, Ryan, Sanson, Shaw, Sparrow, Summerhayes, Terauds and Wall2012b). Despite a high degree of isolation between continental habitats (Convey et al. Reference Convey, Chown, Clarke, Barnes, Bokhorst, Cummings, Ducklow, Frati, Green, Gordon, Griffiths, Howard-Williams, Huiskes, Laybourn-Parry, Lyons, McMinn, Morley, Peck, Quesada, Robinson, Schiaparelli and Wall2014), the distribution patterns of Antarctic species may shift southwards and increasingly overlap, possibly eroding the extensive endemism among many Antarctic species (Nielsen & Wall Reference Nielsen and Wall2013), particularly when considering human-mediated dispersal. Additionally, non-indigenous species may outcompete local endemics in an increasingly accommodating environment, particularly in the sub-Antarctic (Frenot et al. Reference Frenot, Chown, Whinam, Selkirk, Convey, Skotnicki and Bergstrom2005, Hughes & Convey Reference Hughes and Convey2010, Hughes et al. Reference Hughes, Convey, Maslen and Smith2010).
Current Antarctic biology is primarily influenced by the desire to conserve the unique and still largely uncharacterized biodiversity of the continent and surrounding islands. Elucidating distribution patterns of terrestrial communities and identifying biotic elements most vulnerable to climate change have been deemed some of the most important goals of Antarctic biological conservation (Kennicutt et al. Reference Kennicutt, Chown, Cassano, Liggett, Massom, Peck, Rintoul, Storey, Vaughan, Wilson and Sutherland2014). Definition and extension of protected areas in Continental Antarctica, particularly in remote locations, is urgently required (Terauds et al. Reference Terauds, Chown, Morgan, Peat, Watts, Keys, Convey and Bergstrom2012, Shaw et al. Reference Shaw, Terauds, Riddle, Possingham and Chown2014), coupled with increased monitoring of these areas for the introduction of taxa from the sub- and Maritime Antarctic (Chown et al. Reference Chown, Huiskes, Gremmen, Lee, Terauds, Crosbie, Frenot, Hughes, Imura, Kiefer, Lebouvier, Raymond, Tsujimoto, Ware, van de Vijver and Bergstrom2012a, Shaw et al. Reference Shaw, Terauds, Riddle, Possingham and Chown2014, McGeoch et al. Reference McGeoch, Shaw, Terauds, Lee and Chown2015). Efforts to capture heterogeneous patterns in terrestrial biodiversity, and to assess the future impact of alien species, require densely spaced biological and environmental survey data (Shaw et al. Reference Shaw, Terauds, Riddle, Possingham and Chown2014, McGeoch et al. Reference McGeoch, Shaw, Terauds, Lee and Chown2015). The HTS-supported molecular methods are particularly powerful in resolving Antarctic endemics from non-indigenous species that are not easily detected or are difficult to identify (Hughes & Convey Reference Hughes and Convey2012, Chown et al. Reference Chown, Hodgins, Griffin, Oakeshott, Byrne and Hoffmann2015b); such methods could inform, for example, on the number of eukaryotic alien and invasive species per biogeographical region in standardized frameworks (McGeoch et al. Reference McGeoch, Shaw, Terauds, Lee and Chown2015).
Continent-wide survey data and time series monitoring
The HTS-based metabarcoding approaches are regarded as more efficient compared with morphological methods for assessing the ecological integrity and health of diverse marine and terrestrial environments, by providing a uniform, swift and economical means of species identification (Aylagas et al. Reference Aylagas, Borja and Rodríguez-Ezpeleta2014, Drummond et al. Reference Drummond, Newcomb, Buckley, Xie, Dopheide, Potter, Heled, Ross, Tooman, Grosser, Park, Demetras, Stevens, Russell, Anderson, Carter and Nelson2015). Potential applications to Antarctic environments are now being realized, where HTS-based metabarcoding studies similarly offer a simple, cost-efficient workflow and rich sequence information that can be easily combined or re-analysed in more detailed integrative studies (Gutt et al. Reference Gutt, Zurell, Bracegridle, Cheung, Clark, Convey, Danis, David, De Broyer, di Prisco, Griffiths, Laffont, Peck, Pierrat, Riddle, Saucede, Turner, Verde, Wang and Grimm2012, Chown et al. Reference Chown, Hodgins, Griffin, Oakeshott, Byrne and Hoffmann2015b).
In order to use the full potential of HTS for Antarctic biodiversity research and ecology, we suggest i) designing studies with close consideration of research goals defined by the international community (Kennicutt et al. Reference Kennicutt, Chown and Cassano2015), ii) designing studies with larger numbers of samples, for example, across variable spatial and temporal scales, similar to approaches used by Dornelas et al. (Reference Dornelas, Gotelli, McGill, Shimadzu, Moyes, Sievers and Magurran2014) and Howard-Williams et al. (Reference Howard-Williams, Peterson, Lyons, Cattaneo-Vietti and Gordon2006) or contributing towards such efforts, iii) using a variety of DNA sources for analysis, including historical material from museum collections or historical Antarctic voyages (Headland Reference Headland2009), iv) providing well-documented analysis code with all published HTS data, v) further developing laboratory and analysis protocols for metabarcoding and metagenomic approaches suitable to investigate Antarctic habitats, and finally, vi) generating reference DNA sequences for Antarctic species identification using α taxonomic (including morphological) approaches (Turrill Reference Turrill1938).
Summary and conclusions
Metabarcoding analysis of mixed template and environmental DNA is a valuable option to describe the composition and distribution of the cryptic and heterogeneously distributed terrestrial biota of Antarctica. Metabarcoding and metagenomic approaches have proven helpful in describing bacterial and hypolithic communities in ice-free regions of Antarctica and could similarly be applied to many other taxa on the continent, including communities inhabiting snow and ice, as well as lake and marine sediments. In comparison to traditional molecular methods, HTS-based approaches yield large amounts of detailed data with relatively simple and time-efficient laboratory workflows, coupled with straightforward fieldwork. Multiple laboratory developments have recently improved the cost efficiency of PCR-based library generation allowing parallel processing of large sample numbers. Drawbacks of amplicon library generation can be alleviated by alternative library preparation methods. By providing a consistent and efficient means of species identification, as well as insights into the functional diversity of such habitats, HTS-based metabarcoding and metagenomic studies will be a useful tool for assessing the ecological integrity and health of Antarctic habitats. When applied to large sample numbers, across large spatial scales and multiple biota, HTS-based metabarcoding and metagenomic approaches will improve our understanding of Antarctic terrestrial biodiversity on a continental scale.
Acknowledgements
The authors declare no competing interests. The Australian Antarctic Division provided funding under science project 2355 to M.S. The Australian Research Council supported this work through funds from linkage grant LP0991985 to A.C. and M.S. The University of Adelaide supported this project through an International Post-Graduate Research Scholarship to P.C. We thank two anonymous reviewers for helpful comments that improved the manuscript.
Author contribution
P.C. prepared, edited and revised the manuscript. M.S., L.C. and A.C. edited and revised the manuscript.


