Introduction
The mammary gland is a specialized organ in female mammals that evolved for the purpose of milk synthesis and secretion to feed offspring. Due to advancements in genetic selection, however, the lactating dairy cow produces milk volumes that far exceed the neonate's nutritional requirement. As an example, the bovine neonate consumes approximately 11 kg of milk at 30 days of age if allowed ad libitum consumption, whereas the average commercial dairy cow produces on average four times more milk (Jasper and Weary, Reference Jasper and Weary2002). In addition to the remarkable milk volume synthesized by the mammary gland, milk is also rich in nutrients, including protein, carbohydrates, fats, minerals, and vitamins (Haug et al., Reference Haug, Hostmark and Harstad2007). The ability of the mammary gland to synthesize and secrete milk requires a structure that is as unique as its function. In particular, the vascular endothelium is fundamental for the mammary gland to grow and develop and to initiate and sustain milk production. A thin, single layer of mammary capillary endothelial cells forms a semipermeable barrier that facilitates the exchange of serum components to provide oxygen, remove carbon dioxide, and transfer solutes and macromolecules for cellular energy and metabolism (Prosser et al., Reference Prosser, Davis, Farr and Lacasse1996; Andres and Djono, Reference Andres and Djono2010). Endothelial cells also support the robust synthesis and secretion of milk by facilitating a high rate of transfer of blood-derived components, including amino acids and glucose that are necessary for milk production (Aleman et al., Reference Aleman, Lopez, Ordaz, Torres and Tovar2009; Mattmiller et al., Reference Mattmiller, Corl, Gandy, Loor and Sordillo2011). To maintain adequate delivery of milk components and substrates, endothelial cells directly orchestrate vascular tone, blood fluidity, and vascular permeability to support both the vasculature and underlying milk-producing tissue (Prosser et al., Reference Prosser, Davis, Farr and Lacasse1996).
In addition to supporting lactation physiology, the endothelium also plays a direct role in orchestrating host defense to infectious bacterial pathogens. The mammary gland is exposed to outside environment through the teat canal. If bacteria are able to penetrate the teat end barrier, then the efficiency of mammary gland inflammatory responses will determine if mastitis manifests. Vascular endothelial cells play a central role in the facilitating movement of soluble and cellular host defense mechanisms into mammary tissues during the initial stages of bacterial invasion. An efficient inflammatory response will recognize and eliminate invading bacteria promptly without causing any discernable changes to mammary tissues (Aitken et al., Reference Aitken, Corl and Sordillo2011a). When the initial inflammatory response fails to prevent the establishment of bacterial infection, however, clinical and/or chronic mastitis will cause impaired milk yield and quality. Indeed, mastitis costs the US dairy industry an estimated 2 billion dollars a year due to reduced milk production, discarded milk following antibiotic treatment, veterinary costs, and replacement animal costs (National Mastitis Council, 2004). Inadequate inflammatory responses can be attributed to deficiencies in normal endothelial cell functions. Endothelial dysfunction is characterized as facilitating the unregulated accumulation of leukocytes at the site of infection, enhanced leakage of plasma proteins into mammary tissues, and disruption in blood flow that contributes to mammary tissue damage and loss of function (Scalia and Lefer, Reference Scalia and Lefer1998; Knepler et al., Reference Knepler, Taher, Gupta, Patterson, Pavalko, Ober and Hart2001; Cassuto et al., Reference Cassuto, Dou, Czikora, Szabo, Patel, Kamath, Belin De Chantemele, Feher, Romero and Bagi2014). Thus, optimal endothelial cell functions are needed to tightly regulate inflammatory responses and prevent immunopathology during bacterial invasion.
Because endothelial cells play a central role in mammary gland health, knowledge of the delineating factors between a successful self-limiting inflammatory response and immunopathology is critical. This review will cover three major topics that discuss how and why: (1) endothelial cells are necessary for the growth and development of the unique mammary gland; (2) endothelial cells modify their phenotype to orchestrate the initiation and resolution of inflammation; and (3) endothelial dysfunction contributes to immunopathology and subsequent tissue damage. A better understanding of the biological mechanisms regulating endothelial cell function is essential for optimizing mammary gland health and increasing the efficiency of milk production in dairy cattle.
Endothelial cells contribute to the specialized structure of the mammary gland
Mammary gland growth and development
Mammogenesis, the first stage of mammary gland development, culminates in the formation of a complex epithelial and endothelial network. Previous studies show that a disruption in the normal progression of murine mammogenesis hinders the ability of the mammary gland to adequately secrete milk (Rossiter et al., Reference Rossiter, Barresi, Ghannadan, Gruber, Mildner, Fodinger and Tschachler2007). Mammogenesis begins during embryogenesis and continues following the birth of the succeeding neonate, at which time it overlaps with the onset of lactation. Studies on mammogenesis were conducted in rats and mice, and from those studies the development of the bovine mammary endothelium can be inferred (Yasugi et al., Reference Yasugi, Kaido and Uehara1989; Matsumoto et al., Reference Matsumoto, Nishinakagawa, Kurohmaru, Hayashi and Otsuka1992; Abdul Awal et al., Reference Abdul Awal, Matsumoto, Toyoshima and Nishinakagawa1996; Rossiter et al., Reference Rossiter, Barresi, Ghannadan, Gruber, Mildner, Fodinger and Tschachler2007). The mammary endothelium is composed of a thin layer of simple squamous endothelial cells, and along with myoepithelial cells and connective tissue, forms a complex vascular network known as the mammary vasculature. A unique feature of the mammary gland vasculature is the basket-like capillary beds surrounding bronchi-like alveoli clusters as demonstrated in murine models (Yasugi et al., Reference Yasugi, Kaido and Uehara1989). Capillary beds surrounding alveoli maximize the surface area for exchange of oxygen and nutrients. To further facilitate exchange, but also maintain a selective barrier, the endothelial surface is largely continuous with some minor areas of fenestration. A continuous endothelium is an uninterrupted layer of cells that restricts the free movement of large solutes and proteins, but allows for exchange of water and other very small molecules (Levick and Smaje, Reference Levick and Smaje1987; Clough, Reference Clough1991; Michel and Curry, Reference Michel and Curry1999). Fenestrations are small pores in endothelial cells that permit passage of small molecules and proteins. Development of the murine and human mammary endothelium establishes a semipermeable barrier that is primarily continuous with some areas of fenestration, which suggests that the bovine mammary gland maintains a similar structure (Stirling and Chandler, Reference Stirling and Chandler1976; Matsumoto et al., Reference Matsumoto, Nishinakagawa, Kurohmaru, Hayashi and Otsuka1992). To further facilitate nutrient transfer for optimal mammary growth, mammogenesis is characterized by an increase in cell number and surface area to provide a maximal interface for nutrient transfer and milk secretion. In murine models, the increase in surface area of the luminal endothelium occurs early in pregnancy by the formation of microvilli and marginal folds on individual endothelial cells (Matsumoto et al., Reference Matsumoto, Nishinakagawa, Kurohmaru, Hayashi and Otsuka1992). Evidence of increased endothelial surface area by the formation of microvilli and marginal folds, minor areas of fenestrations, and basket-like capillary beds surrounding alveoli suggest that optimal mammary function is unequivocally dependent on mammary structure.
Onset of milk synthesis and secretion
Lactogenesis is the acquisition of functional properties enabling the mammary gland to synthesize and secrete large volumes of milk. While much of the research regarding lactogenesis focuses on the differentiation of epithelial cells, endothelial cells also experience structural and functional changes in an effort to support initial and sustained milk production. For example, the number of mitochondria increases in rat mammary endothelial cells, conceivably for the increased energy required for maintaining blood flow, vascular tone, and regulating transport of components needed for milk synthesis (Abdul Awal et al., Reference Abdul Awal, Matsumoto, Toyoshima and Nishinakagawa1996). Pinocytotic vesicles also increase in rat endothelial cells to support efficient transportation of plasma solutes and molecules, such as glucose (Abdul Awal et al., Reference Abdul Awal, Matsumoto, Toyoshima and Nishinakagawa1996). Recent research demonstrates that several glucose transporter molecules (GLUT) are expressed in the bovine mammary gland, including GLUT1, GLUT3, GLUT4, GLUT5, GLUT8, and GLUT12, named according to their order of discovery. In the bovine mammary gland, the mRNA expression of GLUT1, GLUT8, and GLUT12 increased substantially from late in gestation to early lactation (Zhao and Keating, Reference Zhao and Keating2007). Similarly, the mRNA and protein expression of GLUT1 was significantly increased in mammary tissue from early lactation cows compared to non-lactating cows (Komatsu et al., Reference Komatsu, Itoh, Kushibiki and Hodate2005). Mammary tissue from early-lactation dairy cows exhibited increased GLUT1, which was localized to endothelial cells and epithelial cells, compared to 15 days prior to the onset of lactation (Mattmiller et al., Reference Mattmiller, Corl, Gandy, Loor and Sordillo2011). Though the mechanisms that stimulate increased expression of GLUT1 during late pregnancy and early lactation are unclear, previous research demonstrates that hypoxia in the mammary gland elicits a hypoxia inducible factor-α dependent mechanism that results in GLUT1 upregulation (Shao et al., Reference Shao, Wellman, Lounsbury and Zhao2014). In contrast, the regulation of GLUT4 expression is not clear. Some research suggests GLUT4 increases during late lactation in bovine mammary tissue, whereas others suggest that GLUT4 is not expressed in mammary tissue (Komatsu et al., Reference Komatsu, Itoh, Kushibiki and Hodate2005; Mattmiller et al., Reference Mattmiller, Corl, Gandy, Loor and Sordillo2011). However, the expression of glucose transporters, GLUT1 and GLUT4, by primary bovine mammary endothelial cells (BMEC) was confirmed in an in vitro study (Mattmiller et al., Reference Mattmiller, Corl, Gandy, Loor and Sordillo2011). Because glucose transport is a rate-limiting step in optimal lactation, more investigation into the role of GLUT in mammary endothelial cells are necessary. Nonetheless, the localization of glucose transporters to the endothelium and increased mRNA expression in BMEC from lactating cows suggests endothelial cells play an active role in metabolism and nutrient transfer during lactation.
In conjunction with increased transport molecules, murine models demonstrated increased capillary permeability during early lactation, which would support enhanced transfer of fluids and molecules for milk synthesis (Matsumoto et al., Reference Matsumoto, Kurohmaru, Hayashi, Nishinakagawa and Otsuka1994). To further support nutrient transfer, capillaries with thinner walls were in closer contact with the alveoli during late pregnancy and early lactation compared to late lactation (Matsumoto et al., Reference Matsumoto, Nishinakagawa, Kurohmaru, Hayashi and Otsuka1992). Perhaps most telling about the importance of the mammary vasculature in rats is that the development of the vasculature, measured as number of capillaries per individual lobular ductule, surpassed alveolar lobular ductule development during lactation (Ramirez et al., Reference Ramirez, Lee, Schedin, Russell and Masso-Welch2012). Though additional research is still required in dairy cattle, research in other species support the notion that changes in both structure and optimal function of the bovine vasculature during lactogenesis directly affects the ability of epithelial cells to synthesize milk components.
Mammary gland involution
To support maximal milk production in the next lactation, a period of involution is needed for regeneration of bovine mammary tissues (Holst et al., Reference Holst, Hurley and Nelson1987). Dairy cows milked continuously until calving produced approximately 75% less milk compared to the twin given a 60-day non-milking period prior to calving (Swanson, Reference Swanson1965). Involution across species involves a regression in lobulo-alveoli complexity, decreased cell number, and a loss of milk-synthesizing function (Holst et al., Reference Holst, Hurley and Nelson1987; Walker et al., Reference Walker, Bennett and Kerr1989; Tatarczuch et al., Reference Tatarczuch, Philip and Lee1997; Djonov et al., Reference Djonov, Andres and Ziemiecki2001). Involution in murine, goat, and dairy cow mammary glands suggests epithelial cells experience apoptosis at a very high rate following cessation of milking (Wilde et al., Reference Wilde, Addey, Li and Fernig1997). However, other bovine studies demonstrate very little epithelial cell loss during involution even though alveolar lumen area decreases (Holst et al., Reference Holst, Hurley and Nelson1987). Nonetheless, previous research in lactating goats suggests that induction of epithelial cell apoptosis may be initiated by accumulation of milk in the mammary gland (Quarrie et al., Reference Quarrie, Addey and Wilde1994). Whether regression of the bovine vasculature occurs during involution is not clear, although changes in the endothelium during murine and sheep involution may be used to infer changes in the bovine. In the involuting mouse mammary gland, capillary density decreases to a density similar to early pregnancy (Pepper et al., Reference Pepper, Baetens, Mandriota, Di Sanza, Oikemus, Lane, Soriano, Montesano and Iruela-Arispe2000). Studies in sheep suggest that a decrease in mammary capillary density may be a result of endothelial apoptosis. During involution of the ovine mammary gland, apoptosis of endothelial cells was observed in addition to the presence of apoptotic bodies found within the cytoplasm of healthy endothelial cells (Tatarczuch et al., Reference Tatarczuch, Philip and Lee1997). Interestingly, murine models of involution featured endothelial cell apoptosis that was preceded by epithelial cell apoptosis (Djonov et al., Reference Djonov, Andres and Ziemiecki2001). The timing of endothelial cell apoptosis relative to epithelial apoptosis suggests the endothelial cells may be adapting to or dying from an altered microenvironment. However, the major temporal differences between epithelial and endothelial regression during sheep and rat mammary gland involution was not demonstrated, thus more research is required to understand these processes (Walker et al., Reference Walker, Bennett and Kerr1989; Tatarczuch et al., Reference Tatarczuch, Philip and Lee1997). Degradation of the proteins in the basement membrane also occurs during involution and matrix metalloproteinases (MMP) are largely responsible for this event. Data demonstrate that MMP1 and MMP14 are increased in epithelial cells during late involution, whereas MMP2 was only localized to mammary endothelial cells and only during involution (Rabot et al., Reference Rabot, Sinowatz, Berisha, Meyer and Schams2007). In a human umbilical vein endothelial cell (HUVEC) model, withdrawal of growth factor to induce apoptosis resulted in increased MMP2 expression in apoptotic endothelial cells (Levkau et al., Reference Levkau, Kenagy, Karsan, Weitkamp, Clowes, Ross and Raines2002). Activation of MMP2 in HUVEC enables association of MMP2 with receptors for fibronectin and vitronectin, both glycoproteins responsible for the formation of the extracellular matrix and cell adhesion, suggesting apoptosis of endothelial cells during mammary gland involution may be integrin-mediated (Levkau et al., Reference Levkau, Kenagy, Karsan, Weitkamp, Clowes, Ross and Raines2002). Apoptosis may not be solely responsible for decreased vascular networks in the murine mammary gland where capillary regression was noted in the absence of apoptotic endothelial cells (Djonov et al., Reference Djonov, Andres and Ziemiecki2001). The conflicting research in rodent models and the limited amount of research in involuting bovine mammary glands suggests that more research is needed to better understand the role of endothelial cells in the remodeling process.
Endothelial cells prevent mastitis by orchestrating inflammation
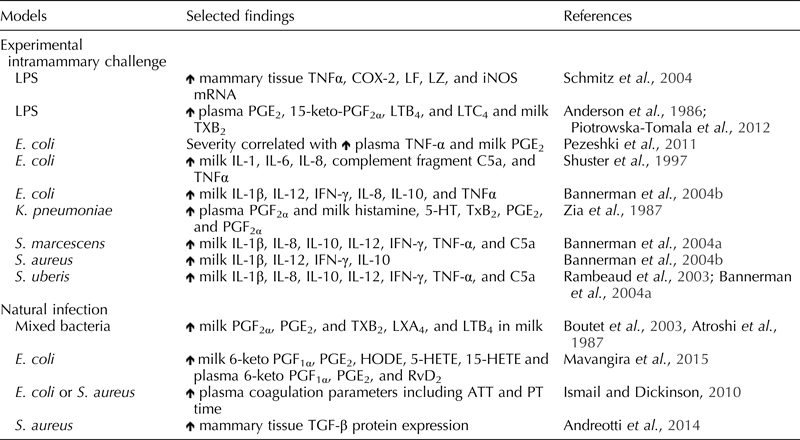
The inflammatory response is a complex, tightly regulated series of events to protect the normal milk-synthesizing function of the bovine mammary gland from mastitis-causing organisms. Endothelial cells regulate a wide variety of homeostatic mechanisms, including maintenance of proper vascular tone, regulation of vascular permeability, modulation of blood fluidity, and regulation of immune responses for protection of the mammary gland during pathogen exposure (Prosser et al., Reference Prosser, Davis, Farr and Lacasse1996; Aitken et al., Reference Aitken, Corl and Sordillo2011a). Exposure of microbes and microbial components initially triggers a response from epithelial cells and resident macrophages to produce and release a variety of inflammatory mediators including cytokines and oxylipids (Table 1). The inflammatory mediators initially recruit phagocytic immune cells and robustly activate endothelial cells. Human and murine models indicate vasodilation of capillaries and increased vascular permeability are necessary for the influx of neutrophils to contain pathogens and limit extravascular tissue damage (Van Nieuw Amerongen and Van Hinsbergh, Reference Van Nieuw Amerongen and Van Hinsbergh2002; Bazzoni and Dejana, Reference Bazzoni and Dejana2004). Endothelial cells also promote active resolution of inflammation to protect the integrity of the vasculature and interstitial tissue (Kadl and Leitinger, Reference Kadl and Leitinger2005). During mastitis, however, the inflammatory response can either be inefficient, excessive, or prolonged, resulting in severe tissue and vascular damage. Thus, it is necessary to differentiate between a normal inflammatory response (self-limiting) and immunopathology (mastitis). The following sections will discuss the change in endothelial cell phenotype needed to orchestrate an efficient inflammatory response.
Table 1. Summary of inflammatory mediator production during bovine mastitis

![]() , Increased; LF, lactoferrin; LZ, lysozyme; LPS, lipopolysaccharide; IL, interleukin; TNF-α, tumor necrosis factor-α; COX-2,cyclooxygenase-2; iNOS, inducible nitric oxide synthase; PG, prostaglandin; LT, leukotriene; TX, thromboxane; IFN-γ, interferon-γ; 5-HT, serotonin; HODE, hydroxyoctadecadienoic acid; HETE, hydroxyeicosatetraenoic acid; RvD, resolvin; ATT, activated partial thromboplastin time; PT, prothrombin time; TGF, transforming growth factor-β.
, Increased; LF, lactoferrin; LZ, lysozyme; LPS, lipopolysaccharide; IL, interleukin; TNF-α, tumor necrosis factor-α; COX-2,cyclooxygenase-2; iNOS, inducible nitric oxide synthase; PG, prostaglandin; LT, leukotriene; TX, thromboxane; IFN-γ, interferon-γ; 5-HT, serotonin; HODE, hydroxyoctadecadienoic acid; HETE, hydroxyeicosatetraenoic acid; RvD, resolvin; ATT, activated partial thromboplastin time; PT, prothrombin time; TGF, transforming growth factor-β.
Endothelial cells modulate changes in vascular tone and blood flow during inflammation
A change in blood flow and vascular tone, characterized by the degree of constriction relative to dilation, is among the initial responses of the vasculature during pathogen exposure (Lacasse et al., Reference Lacasse, Farr, Davis and Prosser1996; Prosser et al., Reference Prosser, Davis, Farr and Lacasse1996; Kobayashi et al., Reference Kobayashi, Oyama, Numata, Rahman and Kumura2013). Myoepithelial cells and endothelial cells work synergistically to either constrict or relax in response to signals primarily produced by endothelial cells (Prosser et al., Reference Prosser, Davis, Farr and Lacasse1996). Endothelial cells produce a variety of vasoactive mediators, such as nitric oxide (NO), prostacyclin (PGI2), endothelin-1, and histamine that function in an autocrine, juxtacrine, and paracrine manner to regulate myoepithelial contraction or relaxation. The most well-studied endothelial-derived regulators of vascular tone during homeostasis are NO and PGI2 in lactating goats, though less is known in the bovine (Nishida et al., Reference Nishida, Harrison, Navas, Fisher, Dockery, Uematsu, Nerem, Alexander and Murphy1992; Nielsen et al., Reference Nielsen, Fleet, Jakobsen and Heap1995; Lacasse et al., Reference Lacasse, Farr, Davis and Prosser1996). At the onset of inflammation, endothelial nitric oxide synthase (eNOS) is activated as a result of increased intracellular calcium. Activation of eNOS metabolizes arginine to citrulline and NO. Subsequently, NO activates guanylyl cyclase to produce cGMP, which inhibits calcium influx into the endothelial cell allowing relaxation of the actin cytoskeleton (Busse and Mulsch, Reference Busse and Mulsch1990). In lactating goats, infusion of the mammary external pudendal artery with an eNOS inhibitor significantly reduces mammary blood flow (Lacasse et al., Reference Lacasse, Farr, Davis and Prosser1996). Mice deficient in eNOS fail to exhibit an early change in the vasculature and the later phase is markedly reduced, suggesting without eNOS activation the inflammatory response cannot be efficiently mounted or proceed normally (Bucci et al., Reference Bucci, Roviezzo, Posadas, Yu, Parente, Sessa, Ignarro and Cirino2005).
Similar to eNOS activation and NO biosynthesis, constitutive cyclooxygenase-1 (COX-1) is activated by an increase in intracellular calcium to facilitate the synthesis of PGI2, an oxylipid. An oxylipid is an oxidized fatty acid and may be synthesized enzymatically or non-enzymatically from many different fatty acids, such as arachidonic acid and linoleic acid. Increased calcium also activates cytosolic phospholipase A2, which cleaves fatty acids from membrane phosphatidylcholine to generate a free fatty acid and lysophosphatidylcholine moiety (Lombardo et al., Reference Lombardo, Fanni, Pluckthun and Dennis1986). During early inflammation, COX-1 oxidizes arachidonic acid to prostaglandin G2 (PGG2), which is quickly reduced to PGH2 by intrinsic peroxidase activity of COX-1 (Fitzpatrick and Soberman, Reference Fitzpatrick and Soberman2001). Next, PGH2 is converted to PGI2 by PGI2 synthase. Activation of the PGI2 receptor, a G-protein-coupled receptor (GPCR), causes an increase in adenylyl cyclase-dependent cAMP and subsequent vasodilation as a result of protein kinase A activation (Murata et al., Reference Murata, Ushikubi, Matsuoka, Hirata, Yamasaki, Sugimoto, Ichikawa, Aze, Tanaka, Yoshida, Ueno, Oh-Ishi and Narumiya1997). Furthermore, sustained PGI2 is dependent on COX-2 and also is activated by increased calcium, but its optimal expression and activity requires transcription and translation induced by pro-inflammatory mediators. Increased expression of COX-2 would aide in sustaining vasodilation during the progression of inflammation. In lactating goats, infusion of the external pudendal artery with PGI2 increased blood flow in the mammary gland, whereas infusion with a PGI2 synthase inhibitor significantly decreased mammary blood flow suggesting the endothelium is sensitive to changes in PGI2 and can alter mammary blood flow (Nielsen et al., Reference Nielsen, Fleet, Jakobsen and Heap1995; Lacasse et al., Reference Lacasse, Farr, Davis and Prosser1996). A change in mammary blood flow following PGI2 and NO exposure suggests that the vasculature in the bovine mammary gland is sensitive to potent vasodilatory mediators. By modulating vascular tone, endothelial cells strive to provide an optimal endothelial surface to facilitate rolling, attachment, and migration of leukocytes to control infection and inflammation.
Though vasodilation is critical to the progression of an appropriate immune response, vasoconstriction during the very early stages of infection and inflammation is protective in the event of mechanical injury and bleeding. Because vasoconstriction limits blood flow, and therefore may compromise the health of the mammary tissue, the release of vasoconstrictors is short-lived and balanced by the sustained release of vasodilators (Prosser et al., Reference Prosser, Davis, Farr and Lacasse1996). Two vasoconstrictors that are rapidly synthesized at the onset of inflammation are platelet-activating factor (PAF) and thromboxane A2 (TXA2). Lysophosphatidylcholine, a lipid moiety generated following the cleavage of arachidonic acid from phosphatidylcholine, acts as a precursor for the production of PAF (Lombardo et al., Reference Lombardo, Fanni, Pluckthun and Dennis1986). Early production of PAF in BMEC and bovine aortic endothelial cells (BAEC) was confirmed following lipopolysaccharide (LPS) exposure (Corl et al., Reference Corl, Gandy and Sordillo2008; Corl et al., Reference Corl, Contreras and Sordillo2010). In murine pulmonary artery endothelial cells, PAF also induced increased production of NO suggesting the presence of a negative feedback loop to prevent sustained vasoconstriction (Predescu et al., Reference Predescu, Knezevic, Bardita, Neamu, Brovcovych and Predescu2013). Another well-known vasoconstrictor produced during inflammation is TXA2, an unstable oxylipid that is degraded to TXB2. The biosynthesis of TXA2 is similar to PGI2 except that thromboxane synthase converts PGH2 to TXA2. Interestingly, BAEC stimulated with a TXA2 mimetic induced the synthesis of the PGI2, further supporting a negative feedback loop established by the production of vasoconstrictors (Clesham et al., Reference Clesham, Parsaee, Joseph, Mcewan and Macdermot1992). In fact, the balance of TXA2 and PGI2 may be more important than absolute concentrations in modulating vascular tone. The production of vasoconstrictors, which also contributes to vasodilator release, suggests that modulation of vascular tone during the initial inflammatory response is tightly regulated to prevent unnecessary damage to blood vessels and interstitial tissue.
In conjunction with changes in vascular tone, reduced blood fluidity also is required during the inflammatory response. During early inflammation, endothelial cells initiate and propagate coagulation by increasing procoagulant properties and decreasing anticoagulants. In BAEC and HUVEC models, thrombus formation is initiated by upregulation of tissue factor expression in response to bacterial toxins and cytokines (Nawroth and Stern, Reference Nawroth and Stern1986; Crossman et al., Reference Crossman, Carr, Tuddenham, Pearson and Mcvey1990; Fei et al., Reference Fei, Berliner, Parhami and Drake1993). The coagulation cascade, initiated by the binding of tissue factor to factor VIIa, acts on factor X to form Factor Xa, which in turn complexes with prothrombin, Factor Va, and calcium to produce thrombin (Nawroth and Stern, Reference Nawroth and Stern1986; Rao and Rapaport, Reference Rao and Rapaport1987). Thrombin then converts fibrinogen to fibrin resulting in the formation of a fibrin clot, thus slowing blood flow at the site of coagulation. Much of the research on the modulation of coagulation in bovine endothelial cells is focused on the effect of cytokines. In BAEC, tumor necrosis factor-α (TNF-α) exposure decreases the expression of thrombomodulin, a major regulatory mechanism of coagulation that prevents the formation of thrombin by generating active protein C (Nawroth and Stern, Reference Nawroth and Stern1986). Further supporting the effect of pro-inflammatory cytokines on BAEC, TNF-α dose-dependently decreased activated protein C, an antioxidant serine protease that inactivates Factor Va and VIIIa (Conway and Rosenberg, Reference Conway and Rosenberg1988). Antithrombin, another anticoagulant factor, binds to heparin-like glycosaminoglycans on the endothelial luminal surface to prevent serine protease activity (Olson et al., Reference Olson, Richard, Izaguirre, Schedin-Weiss and Gettins2010). However, research suggests pro-inflammatory cytokines reduce the expression of heparin sulfate, the major target for antithrombin, by about 50% in porcine aortic endothelial cells (Kobayashi et al., Reference Kobayashi, Shimada and Ozawa1990). Furthermore, thrombin increased cytokine-induced leukocyte migration across a HUVEC monolayer and in an in vivo rabbit dermal inflammation model, supporting the idea that the coagulation cascade is critical for inflammation (Drake et al., Reference Drake, Lopes, Fenton and Issekutz1992). The purpose of coagulation is to reduce blood fluidity to prevent the systemic spread of bacteria and toxins as well as to enhance leukocyte contact with the endothelial cells to facilitate leukocyte migration.
Vascular permeability is altered during inflammation
Endothelial cells mediate the type and quantity of solutes and leukocytes that move back and forth between tissue and blood using selective cellular junctions (Bazzoni and Dejana, Reference Bazzoni and Dejana2004). Perturbation of the endothelium alters endothelial junctions and permeability leading to leakage of plasma proteins and leukocytes. Several endothelial junction proteins can control vascular permeability by regulating cell-to-cell contact and thus forming a highly selective barrier (Blum et al., Reference Blum, Toninelli, Anderson, Balda, Zhou, O'donnell, Pardi and Bender1997; Jiang et al., Reference Jiang, Bryce, Horrobin and Mansel1998). The series of events modulating bovine mammary vascular permeability during inflammation is unknown, but previous studies suggest inflammatory mediators and toxins produced during a normal inflammatory response directly alter endothelial junctions (Bannerman et al., Reference Bannerman, Sathyamoorthy and Goldblum1998; Zhang et al., Reference Zhang, Wang, Gui, Yao, Sun, Wang, Wang, Xie, Yao, Lin and Wu2013). Studies using HUVEC models demonstrate that endothelial tight junctions are loosened by GPCR activation in response to various extracellular mediators, including fatty acids and cytokines (Goeckeler and Wysolmerski, Reference Goeckeler and Wysolmerski1995; Blum et al., Reference Blum, Toninelli, Anderson, Balda, Zhou, O'donnell, Pardi and Bender1997; Jiang et al., Reference Jiang, Bryce, Horrobin and Mansel1998; Michel and Curry, Reference Michel and Curry1999). Tight junctions are composed of transmembrane proteins (e.g., claudins and occludins) and cytosolic proteins (zona occludins), which bridge to cytoskeleton actin filaments. Similarly, in a HUVEC model of inflammation LPS decreased endothelial tight junction proteins concomitant with increased monolayer permeability (Zhang et al., Reference Zhang, Wang, Gui, Yao, Sun, Wang, Wang, Xie, Yao, Lin and Wu2013). Claudin-5 (a tight junction protein) deficient mice exhibited a leaky blood–brain barrier with larger molecules able to move across this extremely restrictive barrier (Nitta et al., Reference Nitta, Hata, Gotoh, Seo, Sasaki, Hashimoto, Furuse and Tsukita2003). Unrestricted movement of macromolecules may contribute to destruction of extravascular tissue and allow systemic infection. Tight junctions are not the only cell connections altered during inflammation. Exposure of bovine pulmonary artery endothelial cells to LPS destabilized endothelial adherens junctions by cleaving β-catenin, thereby disrupting the anchor to the cell cytoskeleton (Bannerman et al., Reference Bannerman, Sathyamoorthy and Goldblum1998). Additionally, blocking VE-cadherin binding in HUVEC increased permeability to solutes and leukocytes suggesting any modulation in endothelial junction expression may severely alter the course of inflammation (Hordijk et al., Reference Hordijk, Anthony, Mul, Rientsma, Oomen and Roos1999). Thus, without proper organization and function of tight junctions and adherens junctions in the mammary endothelium, exchange of solutes and cells is dysregulated and may contribute to tissue damage due to accumulation of plasma components. However, some rearrangement or modulation of endothelial junctions must occur to facilitate effective clearance of mastitis-causing pathogens by phagocytic immune cells.
In dairy cows, most research on barrier permeability is focused on epithelial cells without considering the important contributions of endothelial cells. Early studies suggested that an increase in sodium and chloride in milk from cows with mastitis was indicative of epithelial barrier disruption (Linzell and Peaker, Reference Linzell and Peaker1972). However, researchers also appreciated the presence of serum albumin in milk as indicative of increased permeability at the blood–milk barrier (Kobayashi et al., Reference Kobayashi, Oyama, Numata, Rahman and Kumura2013). Endothelial cells and associated endothelial junctions directly contribute to changes in milk components, such as albumin. Albumin is one of the most important proteins that regulate oncotic pressure and disruption of oncotic pressure typically causes edema in interstitial tissue (Rippe et al., Reference Rippe, Kamiya and Folkow1979). Additionally, albumin can bind many different molecules and act as a chaperone across the endothelium (Spector, Reference Spector1975). Thus, increased albumin in secretory products, i.e., milk, would indicate radical changes at the endothelium. A similar occurrence is demonstrated during early involution, when albumin concentration in milk increases (Poutrel et al., Reference Poutrel, Caffin and Rainard1983; Breau and Oliver, Reference Breau and Oliver1985). The increase in serum albumin may be related to a minimal increase in vascular permeability to support passive immunoglobulin transfer during early involution. In contrast, the increased concentration of serum albumin in early involution may simply be an artifact of decreased milk production with no appreciable change yet in plasma protein transfer (Poutrel et al., Reference Poutrel, Caffin and Rainard1983). Understanding how the endothelium regulates vascular permeability is necessary for discovering new and innovative therapies to prevent systemic infection, as well as add insight to the complex changes that occur during mammary regression.
Leukocyte movement increases across the mammary endothelium during inflammation
The movement of leukocytes into the infected tissue is critical to prevent the progression of mastitis. Leukocytes present in the blood stream are not innately adhesive, but do interact with the endothelium. Under basal conditions neutrophils roll along the vascular barrier and communicate through random and reversible interactions with neutrophil surface selectins. Neutrophil leukocyte–selectin (neutrophil associated; L-selectin) association with the murine endothelium allows contact, but not tethering, capture, or firm adhesion (Kaplanski et al., Reference Kaplanski, Farnarier, Tissot, Pierres, Benoliel, Alessi, Kaplanski and Bongrand1993; Ley et al., Reference Ley, Bullard, Arbones, Bosse, Vestweber, Tedder and Beaudet1995). The purpose of neutrophil rolling is to survey the vasculature for cytokines, oxylipids, or reactive oxygen species (ROS) signaling microbial infection (Ley et al., Reference Ley, Bullard, Arbones, Bosse, Vestweber, Tedder and Beaudet1995). The production of ROS is increased during early inflammation due to increased adenosine triphosphate (ATP) production through the electron transport chain and as a result of respiratory burst by phagocytic cells (Parnham et al., Reference Parnham, Bittner and Leyck1987; Aon et al., Reference Aon, Stanley, Sivakumaran, Kembro, O'rourke, Paolocci and Cortassa2012). At the initiation of inflammation, activation of GPCRs induces the release of pre-synthesized platelet-selectin (P-selectin) from specialized endothelial organelles called Weibel–Palade bodies as demonstrated in HUVEC (McEver et al., Reference Mcever, Beckstead, Moore, Marshall-Carlson and Bainton1989). Binding of P-selectin to leukocyte-associated P-selectin glycoprotein ligand-1 forms a tight, but reversible, attachment between endothelial cells and leukocytes. In BMEC, the mRNA expression of P-selectin is increased following exposure to TNF-α and in P-selectin deficient mice, rolling of neutrophils on the endothelial surface and neutrophil migration are reduced, illustrating the vital role of initial P-selectin binding in leukocyte migration (Mayadas et al., Reference Mayadas, Johnson, Rayburn, Hynes and Wagner1993; Aitken et al., Reference Aitken, Corl and Sordillo2011b). Capture, but not firm adhesion, of neutrophils is complete with endothelial-selectin (E-selectin), but its transcription and expression requires gene transcription and translation. In both BMEC and HUVEC, increased temporal synthesis and expression of P-selectin and E-selectin is the result of various inflammatory mediators, including cytokines and ROS (Scholz et al., Reference Scholz, Devaux, Hirche, Potzsch, Kropp, Schaper and Schaper1996; Maddox et al., Reference Maddox, Aherne, Reddy and Sordillo1999; Aitken et al., Reference Aitken, Corl and Sordillo2011b). Stimulation of BMEC with TNF-α induced an increase in E-selectin expression at 3 and 6 h, whereas P-selectin expression was only significantly increased at 3 h (Maddox et al., Reference Maddox, Aherne, Reddy and Sordillo1999). The time-dependent expression of adhesion molecules involved in tethering and capture during inflammation further supports the temporal, cooperative effort between P- and E-selectin that must exist in the bovine mammary gland. The contribution of endothelial cells to tethering and capture of neutrophils during inflammation is unequivocal and necessary for an appropriate, timely response.
Firm adhesion follows capture of leukocytes and is necessary for diapedesis of leukocytes into the inflamed tissue. Firm adhesion is achieved by neutrophil ligand binding to intercellular adhesion molecule-1 (ICAM-1), ICAM-2, and vascular cell adhesion molecule-1 (VCAM-1) (Sans et al., Reference Sans, Panes, Ardite, Elizalde, Arce, Elena, Palacin, Fernandez-Checa, Anderson, Lobb and Pique1999). Macrophage-1 antigen binds to endothelial ICAM-1, whereas lymphocyte function associated antigen-1 preferentially binds ICAM-2. In vivo data during bovine mammary gland inflammation are limited regarding firm adhesion, but murine and human studies provide information about endothelial cells in orchestrating leukocyte adhesion and migration (Sans et al., Reference Sans, Panes, Ardite, Elizalde, Arce, Elena, Palacin, Fernandez-Checa, Anderson, Lobb and Pique1999; Hafezi-Moghadam et al., Reference Hafezi-Moghadam, Noda, Almulki, Iliaki, Poulaki, Thomas, Nakazawa, Hisatomi, Miller and Gragoudas2007). Very late antigen-4 (VLA-4) is the ligand for VCAM-1, and when VLA-4 is blocked on rat leukocytes, adhesion to the endothelium is significantly reduced (Hafezi-Moghadam et al., Reference Hafezi-Moghadam, Noda, Almulki, Iliaki, Poulaki, Thomas, Nakazawa, Hisatomi, Miller and Gragoudas2007). In a rat model of colitis, blockade of ICAM-1 slightly reduced leukocyte adhesion, but blockade of VCAM-1 prevented all adhesion of leukocytes to colonic venules suggesting VCAM-1 may be the predominant molecule in firm adhesion (Sans et al., Reference Sans, Panes, Ardite, Elizalde, Arce, Elena, Palacin, Fernandez-Checa, Anderson, Lobb and Pique1999). In bovine in vitro models, several studies demonstrated the ability to increase the expression of ICAM-1 and VCAM-1 following exposure to TNF-α and PAF (Sordillo et al., Reference Sordillo, Weaver, Cao, Corl, Sylte and Mullarky2005; Corl et al., Reference Corl, Gandy and Sordillo2008). Additionally, a mixture of saturated and unsaturated fatty acids increased the expression of ICAM-1 in BAEC (Aitken et al., Reference Aitken, Corl and Sordillo2011b; Contreras et al., Reference Contreras, Mattmiller, Raphael, Gandy and Sordillo2012a, Reference Contreras, Raphael, Mattmiller, Gandy and Sordillob). In contrast, omega-3 fatty acids reduced ICAM-1 and VCAM-1 expression in cytokine-stimulated human endothelial cells (De Caterina et al., Reference De Caterina, Cybulsky, Clinton, Gimbrone and Libby1994). Furthermore, arachidonic acid-derived 15-hydroperoxyeicosatetraenoic acid (15-HPETE) and linoleic acid-derived 13-hydroperoxyoctadecadienoic acid, both produced early in the inflammatory response, induced an increase in BAEC and HUVEC ICAM-1 expression at low doses (Friedrichs et al., Reference Friedrichs, Toborek, Hennig, Heinevetter, Muller and Brigelius-Flohe1999; Sordillo et al., Reference Sordillo, Streicher, Mullarky, Gandy, Trigona and Corl2008). Rapid biosynthesis of oxylipids supports robust and acute expression of endothelial adhesion molecules to expedite neutrophil migration into the infected tissue. The sensitivity of the endothelium to fatty acids and oxylipids is important because it suggests that the inflammatory response may be modulated with only small changes in autocrine, juxtacrine, and paracrine mediators.
Though research focusing on the process of leukocyte migration is limited, there is no doubt that endothelial–leukocyte cooperation is necessary to successfully bring neutrophils to the site of infection. Platelet–endothelial cell adhesion molecule-1 (PECAM-1) is thought to be the primary molecule that mediates migration of leukocytes between across the endothelium through the formation of homodimers between endothelial cells and neutrophils (Schenkel et al., Reference Schenkel, Mamdouh, Chen, Liebman and Muller2002). Though there was no change in BMEC PECAM-1 mRNA expression following TNF-α exposure, previous human studies showed that blocking neutrophil PECAM-1 or HUVEC prevents transmigration, suggesting without PECAM-1 binding an efficient inflammatory response would not occur (Muller et al., Reference Muller, Weigl, Deng and Phillips1993; Aitken et al., Reference Aitken, Corl and Sordillo2011b). Moreover, expression of PECAM-1 was upregulated in an in vivo murine model following exposure to cytokines, such as IL-1β and TNF-α, (Thompson et al., Reference Thompson, Noble, Larbi, Dewar, Duncan, Mak and Nourshargh2001). Additionally, PECAM-1 receptors may rearrange and move to junctional complexes to support leukocyte migration. For example, TNF-α and IFN-γ induced PECAM-1 redistribution on the HUVEC (Romer et al., Reference Romer, Mclean, Yan, Daise, Sun and Delisser1995). Expression of PECAM-1 during bovine mammary inflammation is not well characterized and requires further exploration. However, based on other human and murine inflammatory models, it is appreciated that an optimal inflammatory response requires a coordinated effort between endothelial cells and surrounding cell types to facilitate neutrophil migration to the site of inflammation.
Resolution of inflammation is necessary to prevent vascular and mammary tissue damage
The process of active resolution is not completely understood, but current research suggests that there is a switch from a pro-inflammatory to an anti-inflammatory/pro-resolving cellular phenotype (Serhan et al., Reference Serhan, Chiang and Van Dyke2008a). A proper inflammatory response is self-limiting with the final phase being resolution. The sequence of events that lead to resolution of inflammation and tissue repair is not well defined due to the involvement of many different cells and pathways. Thus, the topics and order of discussion for the following endothelial mechanisms is based on their hierarchal importance in reducing leukocyte migration and promoting tissue repair.
Endothelial cells are sensitive to ROS and free radicals, collectively called pro-oxidants, and during inflammation there is increased pro-oxidant production surrounding and within endothelial cells (Zweier et al., Reference Zweier, Broderick, Kuppusamy, Thompson-Gorman and Lutty1994). Intracellular pro-oxidants are derived from the mitochondrial electron transport chain during ATP production and extracellular pro-oxidants are predominantly derived from nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity during phagocytic respiratory burst (Karlsson et al., Reference Karlsson, Markfjall, Stromberg and Dahlgren1995; Aon et al., Reference Aon, Stanley, Sivakumaran, Kembro, O'rourke, Paolocci and Cortassa2012). The continued presence of pro-oxidants can contribute to the development of oxidative stress. Oxidative stress is an imbalance of antioxidant mechanisms and pro-oxidant molecules resulting in damage to cellular lipids, proteins, and DNA (Sordillo et al., Reference Sordillo, Contreras and Aitken2009). To mediate resolution, antioxidants must be produced to counteract the damaging effects of pro-oxidants. The most well-studied enzymatic antioxidant defenses include glutathione peroxidase (GPx), superoxide dismutase (SOD), and thioredoxin reductase (TrxR) (Bowler et al., Reference Bowler, Nicks, Tran, Tanner, Chang, Young and Worthen2004; Aon et al., Reference Aon, Stanley, Sivakumaran, Kembro, O'rourke, Paolocci and Cortassa2012). Superoxide dismutase either adds or removes an electron from O2 − to form O2 or H2O2, which will be catalyzed to oxidized glutathione and H2O by GPx (Bowler et al., Reference Bowler, Nicks, Tran, Tanner, Chang, Young and Worthen2004). The in vivo action of TrxR, GPx, and SOD is unknown during self-limiting bovine mammary gland inflammation, but in vitro models in bovine, murine, and human endothelial cells provide an opportunity to discuss how the lack of antioxidants defenses promotes inflammation. The overexpression of GPx-1 protected BAEC-derived NO production in an endothelial injury model (Weiss et al., Reference Weiss, Zhang, Heydrick, Bierl and Loscalzo2001). Conversely, deficiency of GPx-1 during TNF-α stimulation in human microvascular endothelial cells exacerbated the expression of VCAM-1 and ICAM-1 and subsequently enhanced ROS production (Lubos et al., Reference Lubos, Kelly, Oldebeken, Leopold, Zhang, Loscalzo and Handy2011). Mice deficient in extracellular endothelial-derived SOD demonstrated significantly exaggerated lung inflammation characterized by robust and sustained pro-inflammatory cytokine production (Bowler et al., Reference Bowler, Nicks, Tran, Tanner, Chang, Young and Worthen2004). Importantly, neutrophil infiltration was persistent in SOD knockout mice, which was consistent with sustained endothelial adhesion molecule expression, suggesting clearance of pro-oxidants by endothelial cells is critical for decreased leukocyte migration and resolution of inflammation (Bowler et al., Reference Bowler, Nicks, Tran, Tanner, Chang, Young and Worthen2004).
While GPx and SOD directly reduce ROS and free oxygen radicals, TrxR reduces oxidized thioredoxin, so that it may reduce other pro-oxidants (Trigona et al., Reference Trigona, Mullarky, Cao and Sordillo2006). Following low-dose H2O2 exposure in HUVEC, thioredoxin mRNA and protein expression were significantly increased indicating, its role in antioxidant defense (Haendeler et al., Reference Haendeler, Tischler, Hoffmann, Zeiher and Dimmeler2004). Interestingly, TrxR can also reduce GPx in human plasma after pro-oxidant quenching suggesting redundancy in antioxidant defense system (Bjornstedt et al., Reference Bjornstedt, Xue, Huang, Akesson and Holmgren1994). However, it is unknown whether this occurs in the bovine mammary gland or in the mammary endothelial cells. Overall these studies suggest that for a self-limiting inflammatory response to occur, adequate antioxidants and antioxidant enzymes must be available to counteract increased pro-oxidants during inflammation.
Another new and burgeoning area of research that involves active resolution of inflammation is the production of anti-inflammatory and pro-resolving lipid mediators. The major classes of anti-inflammatory and pro-resolving oxylipids include resolvins (RV), protectins (PD), and lipoxins (LX), which are synthesized from fatty acids. The RV are synthesized from eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA), and PD are derived from DHA. Resolvins derived from EPA are enzymatically formed by the sequential oxidation of cytochrome P450 and 5-LOX, whereas DHA-derived RV (RvD) are generally derived by the sequential oxidation 15-LOX and 5-LOX, similar to AA-derived LX. On the other hand, PD is formed by 15-LOX oxidation and subsequent hydrolysis. There is not a great deal of in vivo bovine research available for review in this new area, however, research in in vitro bovine models demonstrate the anti-inflammatory effects of increased EPA and DHA on cellular phenotype and subsequent oxylipid production. For example, in BAEC increased omega-3 fatty acid content (DHA and EPA) reduced pro-inflammatory cytokine expression, adhesion molecule expression, ROS production, and pro-inflammatory oxylipid production (Contreras et al., Reference Contreras, Mattmiller, Raphael, Gandy and Sordillo2012a). Also, the production of DHA-derived oxylipids, including RvD and PD, and AA-derived LXA4, were increased in BAEC exposed to increased omega-3 fatty acids (Contreras et al., Reference Contreras, Mattmiller, Raphael, Gandy and Sordillo2012a). In a HUVEC model of inflammation, RvD1 prevented the reorganization of tight junction proteins and abrogated any changes in vascular permeability induced by LPS (Zhang et al., Reference Zhang, Wang, Gui, Yao, Sun, Wang, Wang, Xie, Yao, Lin and Wu2013). Similar to RV function, PD also has the ability to promote endothelial barrier function. In human microvascular endothelial cells, PD1 effectively prevented the LTB4-induced migration of neutrophils (Serhan et al., Reference Serhan, Gotlinger, Hong, Lu, Siegelman, Baer, Yang, Colgan and Petasis2006). In addition, PD1 administration also was able to abrogate the inflammatory response after induction of murine peritonitis (Serhan et al., Reference Serhan, Gotlinger, Hong, Lu, Siegelman, Baer, Yang, Colgan and Petasis2006). Thus, research suggests that administration of omega-3-derived oxylipids may be beneficial in preventing immunopathogenesis; however, the complexity of oxylipid biosynthesis necessitates further research in human and veterinary species.
Oxylipids derived from omega-6 fatty acids may also play a very important role in protecting the endothelial barrier. Arachidonic acid-derived LX are primarily synthesized with the cooperative effort of both 5-LOX and 15-LOX (Serhan et al., Reference Serhan, Yacoubian and Yang2008b). Lipoxins inhibit chemotaxis and migration of neutrophils, reduce ROS production, and prevent activation of nuclear factor-κB (NF-κB) (Serhan et al., Reference Serhan, Chiang and Van Dyke2008a). In the context of endothelial barrier integrity, LXA4 prevents LPS- or PAF-induced increased vascular permeability suggesting LXA4 acts as an anti-inflammatory protector of the endothelium (Ereso et al., Reference Ereso, Cureton, Cripps, Sadjadi, Dua, Curran and Victorino2009). In addition, LXA4 induces IL-10 production in a HUVEC model while concomitantly inhibiting production of pro-inflammatory cytokines and vascular adhesion molecules (Baker et al., Reference Baker, O'meara, Scannell, Maderna and Godson2009). Consequently, active resolution is dependent on the contribution of potent anti-inflammatory oxylipids and given their ability to modulate many aspects of inflammation warrants continued research in human and veterinary species.
As oxylipids may directly modulate cytokine production to promote resolution, other negative feedback loops and checkpoints also exist to prevent sustained inflammation. Cytokine expression, as a result of NF-κB signaling, is a major pro-inflammatory pathway activated following the detection of bacterial pathogens (Nadjar et al., Reference Nadjar, Tridon, May, Ghosh, Dantzer, Amedee and Parnet2005). Thus, it makes sense that NF-κB signaling should be tightly regulated to prevent sustained activation. One such checkpoint is inhibition of NF-κB signaling pathway by inhibitor of κB (IκB). The inhibitor IκBα prevents NF-κB binding, and thus prevents activation of pro-inflammatory gene expression. Though IκBα is degraded at the onset of inflammation, sustained activation of NF-κB initiates the expression of IκB, which acts as a negative feedback loop (De Martin et al., Reference De Martin, Vanhove, Cheng, Hofer, Csizmadia, Winkler and Bach1993). Additionally, IκBα may directly decrease the expression of IL-1, IL-6, IL-8, and VCAM in porcine aortic endothelial cells (Wrighton et al., Reference Wrighton, Hofer-Warbinek, Moll, Eytner, Bach and De Martin1996). Aside from pro-inflammatory cytokines, endothelial cells also produce anti-inflammatory cytokines, including IL-10, IL-1 receptor agonist, and transforming growth factor-β (TGF-β), which decrease the expression of pro-inflammatory cytokines and also promote tissue repair (Cromack et al., Reference Cromack, Porras-Reyes, Purdy, Pierce and Mustoe1993). In HUVEC, exposure to TGF-β decreased both baseline E-selectin and TNF-α-stimulated E-selectin mRNA and protein expression by more than 50% (Gamble et al., Reference Gamble, Khew-Goodall and Vadas1993). Though the mechanism by which tissue repair is initiated remains unclear, TGF-β contributes substantially through activation of angiogenesis and induction of matrix component synthesis (Ignotz and Massague, Reference Ignotz and Massague1986; Ferrari et al., Reference Ferrari, Cook, Terushkin, Pintucci and Mignatti2009). Decreased expression of pro-inflammatory cytokines, in conjunction with increased anti-inflammatory cytokines and growth factors, is necessary for controlling chronic inflammation and immunopathology.
Endothelial dysfunction as an underlying cause for immunopathology
Bovine mastitis results when there is not a self-limiting inflammatory response and may be a result of endothelial cell dysfunction. Endothelial dysfunction can contribute to an aberrant inflammatory response as a result of breakdown of tight junction proteins, loss of barrier integrity, increased attachment of leukocytes and platelets, and in the most severe cases, leading to sepsis and tissue hypoperfusion (Levi et al., Reference Levi, Van Der Poll, Ten Cate and Van Deventer1997; Van Nieuw Amerongen and Van Hinsbergh, Reference Van Nieuw Amerongen and Van Hinsbergh2002; Winn and Harlan, Reference Winn and Harlan2005). Based on previous in vitro BMEC and BAEC studies, as well as research in other species and disease models as a reference, it is possible to speculate how endothelial cell dysfunction can contribute to immunopathology during mastitis (Fig. 1). The following sections discuss the impact of sustained inflammation and accumulation of pro-oxidants on endothelial function and survival.

Fig. 1. (a) During homeostasis, endothelial cells maintain adequate vascular tone, permeability, and leukocyte migration. (b) At the onset of infection-induced inflammation, endothelial cells acquire new properties to facilitate clearance of the bacteria. Activated endothelial cells express adhesion molecules and produce increased concentrations of ROS, vasoactive lipid mediators, and cytokines. Increased vasodilation and chemokines elicit neutrophils to adhere to and cross the endothelium. Some endothelial cells may exhibit morphological changes during inflammation, those of which are eliminated during the resolution and wound healing phase of inflammation. (c) An unregulated immune response results in prolonged activation of endothelial cells and accumulation of activated neutrophils at the endothelial surface causing irreversible cell damage. Oxidative stress as a result of accumulated ROS, free radicals, and lipid hydroperoxides further contributes to cellular damage and apoptosis of endothelial cells.
Oxidative stress negatively affects antioxidant pathways and oxylipid biosynthesis
Oxidative stress contributes to immunopathology by modulating endothelial function. During inflammation, endothelial metabolism is increased to support a variety of mechanisms, including increased adhesion molecule expression, cytokine expression, and antioxidant pathways (Kadl and Leitinger, Reference Kadl and Leitinger2005). Functional roles of antioxidant systems may be decreased due to quenching by existing ROS and free radicals, down regulation of transcriptional expression, and inactivation of antioxidant enzymes (Blum and Fridovich, Reference Blum and Fridovich1985). In BAEC, reduced TrxR activity during oxidative stress resulted in decreased heme oxygenase-1 (HO-1), an enzyme that catalyzes the degradation of ROS-inducing free heme (Trigona et al., Reference Trigona, Mullarky, Cao and Sordillo2006; Fortes et al., Reference Fortes, Alves, De Oliveira, Dutra, Rodrigues, Fernandez, Souto-Padron, De Rosa, Kelliher, Golenbock, Chan and Bozza2012). Decreased HO-1 would exacerbate oxidative stress and potentially result in endothelial cell death. In fact, pro-apoptotic markers were increased during oxidative stress in BAEC, a finding that was correlated to decreased HO-1 and TrxR (Trigona et al., Reference Trigona, Mullarky, Cao and Sordillo2006). In vitro BMEC models shed further light on how oxidative stress affects cellular function eventually leading to endothelial dysfunction. Endothelial cells (BMEC and BAEC) increased production of TXB2 and PAF, and decreased production of PGI2 during oxidative stress potentially compromising maintenance of vascular tone (Cao et al., Reference Cao, Reddy and Sordillo2000; Weaver et al., Reference Weaver, Maddox, Cao, Mullarky and Sordillo2001). Additionally, changes in endothelial cell metabolism during oxidative stress resulted in the accumulation of bioactive oxylipids, such as 15-HPETE (Weaver et al., Reference Weaver, Maddox, Cao, Mullarky and Sordillo2001). Arachidonic acid-derived 15-HPETE is a lipid hydroperoxide derived by 15-LOX oxidation and can have similar effects on endothelial cell health as ROS. Such changes in oxylipid biosynthesis during oxidative stress can cause apoptosis of bovine endothelial cells, which may lead to disruption of the endothelial barrier (Sordillo et al., Reference Sordillo, Weaver, Cao, Corl, Sylte and Mullarky2005). Our understanding of these mechanisms during bovine mastitis is less clear; however, in vitro reports support the contention that oxidative stress can compromise endothelial integrity.
Oxidative stress contributes to decreased NO
In human diseases, endothelial dysfunction is classically characterized as the reduced availability of NO resulting in altered vascular tone and blood flow. The availability of NO ensures that the mammary vasculature remains relaxed, as opposed to being in a state of vasoconstriction (Ohashi et al., Reference Ohashi, Kawashima, Hirata, Yamashita, Ishida, Inoue, Sakoda, Kurihara, Yazaki and Yokoyama1998). Chemical inhibition of eNOS in HUVEC induces intracellular oxidative stress contributing to increased adhesion of neutrophils (Niu et al., Reference Niu, Smith and Kubes1994). During a dysfunctional inflammatory response, NO levels may be reduced dramatically as a result of several different mechanisms. First, reduced NO may be a function of eNOS uncoupling. Uncoupling of eNOS occurs when available O2 is oxidized to form superoxides preventing its combination with L-arginine to generate NO (Vasquez-Vivar et al., Reference Vasquez-Vivar, Kalyanaraman, Martasek, Hogg, Masters, Karoui, Tordo and Pritchard1998). In addition to reduced NO synthesis, superoxide, generated during oxidative stress, is toxic to endothelial cells and other cells if not reduced by superoxide dismutase. Aside from superoxide production by eNOS uncoupling, activated neutrophils arrested at the endothelial barrier produce superoxide and other ROS/free radicals by the action of NADPH oxidase (Karlsson et al., Reference Karlsson, Markfjall, Stromberg and Dahlgren1995; Aon et al., Reference Aon, Stanley, Sivakumaran, Kembro, O'rourke, Paolocci and Cortassa2012). The excess ROS further uncouples eNOS by oxidizing BH4, a cofactor for eNOS activity (Xia et al., Reference Xia, Tsai, Berka and Zweier1998). Though eNOS activation or uncoupling during bovine mastitis is unknown, accumulation of neutrophils at the endothelial interface was demonstrated during Streptococcus uberis mastitis along with destruction of the surrounding tissue (Thomas et al., Reference Thomas, Haider, Hill and Cook1994). Levels of NO also may be reduced by ROS consumption. Accumulation of activated neutrophils would allow for increased ROS production, such as superoxide, at the endothelial surface, which would be able to act on NO directly rather than affecting eNOS activity. Peroxynitrite, formed by the reaction of NO and superoxide, induced barrier dysfunction, endothelial protein oxidation, and cytoskeleton rearrangement in porcine pulmonary artery endothelial cells (Knepler et al., Reference Knepler, Taher, Gupta, Patterson, Pavalko, Ober and Hart2001). Thus, reduced NO through eNOS uncoupling or quenching by oxygen radicals prevents endothelial cells from maintaining proper vascular tone and perpetuates a dysfunctional endothelial phenotype.
Prolonged or excessive inflammation induces apoptosis of endothelial cells
The end result of persistent endothelial dysfunction is apoptosis, which may be activated through both intrinsic and extrinsic pathways. Apoptosis of endothelial cells would compromise the mammary endothelium thereby disrupting the semi-permeable endothelial barrier. The extrinsic pathway is initiated through receptor activation by death ligands, including TNF-α and LPS. Exposure of BMEC to TNF-α induced a pro-apoptotic response, suggesting cytokines meant to activate endothelial cells to facilitate pathogen clearance may also damage the gatekeeper cells at the blood–milk barrier (Aitken et al., Reference Aitken, Corl and Sordillo2011b). Furthermore, apoptosis was documented during oxidative stress in a BAEC model supporting the intrinsic pathway of apoptosis (Sordillo et al., Reference Sordillo, Weaver, Cao, Corl, Sylte and Mullarky2005; Trigona et al., Reference Trigona, Mullarky, Cao and Sordillo2006). The intrinsic pathway is activated by cellular DNA damage and/or accumulation of intracellular ROS, which causes mitochondrial damage triggering apoptosis. During bovine mastitis, ROS and free radicals accumulate at the endothelial barrier due to the activated nature of endothelial cells and activated neutrophils that are migrating or immobilized at the barrier (Thomas et al., Reference Thomas, Haider, Hill and Cook1994). Previous HUVEC models demonstrate that oxygen radicals activate protein kinase D and ASK-1/JNK, which activates the JNK/p38 MAPK apoptosis pathway (Zhang et al., Reference Zhang, Zheng, Storz and Min2005). Additionally, a HUVEC model demonstrated that apoptotic endothelial cells possess procoagulant and prothrombotic properties, including increased platelet adhesion and decreased thrombomodulin, heparin sulfate, and tissue factor pathway inhibitor (Bombeli et al., Reference Bombeli, Karsan, Tait and Harlan1997, Reference Bombeli, Schwartz and Harlan1999). Furthermore, caspase cleavage during apoptosis directly disrupts adherens junctions, allowing the transfer of plasma proteins and fluid in bovine pulmonary artery endothelial cells (Bannerman et al., Reference Bannerman, Sathyamoorthy and Goldblum1998). During bovine mastitis, it seems likely that an inability to rectify the inflammatory response may be due to damaged and apoptotic endothelial cells. Unfortunately, it is unclear when the inflammatory response can be targeted to prevent destruction of the bovine mammary vascular endothelium and represents a substantial gap in our current knowledge.
Conclusions
Endothelial cells are no longer viewed as static barriers at the interface between blood and tissue. Endothelial cells regulate transfer of solutes and macromolecules, leukocyte migration, vascular tone, and blood flow during homeostasis and inflammation. The ability of a thin layer of endothelial cells to serve such a prominent role is directly related to their proximity to the circulation and to the underlying interstitial tissue. Specifically, capillaries encircling milk-producing alveoli in the bovine mammary gland offer increased surface area for the principle functions of lactation and immune surveillance. However, research is still limited outlining the role of endothelial cells in initiation and resolution of mammary inflammation. Through modulation of vascular tone and blood fluidity, vascular permeability, and endothelial adhesiveness, the endothelium enables the progression of a self-limiting inflammatory response to support clearance of the pathogen and protect the milk-producing tissue. However, mediators that are important for progression of mastitis and clearance of pathogens may also be responsible for damage to alveoli in the mammary gland. In particular, oxidative stress is especially detrimental to endothelial function and survival. Thus, understanding how endothelial activation becomes endothelial dysfunction represents a point of potential therapeutic intervention, e.g, preventing or ameliorating oxidative stress. Future studies should focus on the mechanisms that initiate the switch from activation to resolution. Such research would be important to modify exacerbated and sustained inflammatory responses that contribute to inflammatory-based diseases such as bovine mastitis.
Acknowledgments
This work is supported by the National Institute of Food and Agriculture, U.S. Department of Agriculture, under Agreement No. 2015-67011-23012 and Agreement No. 2011-67015-30179.